Le syndrome LPAC (Low Phospholipid-Associated Cholelithiasis) : mythe ou réalité ?
Objectifs pédagogiques
- Connaître la définition et la fréquence du syndrome LPAC
- Savoir évoquer et confirmer un syndrome LPAC
- Connaître le traitement du syndrome LPAC
Habituellement, les calculs biliaires se forment dans la vésicule biliaire et résultent de la précipitation du cholestérol en excès dans la bile. Le plus souvent la lithiase biliaire survient chez des patients en surpoids, de plus de 40 ans et les symptômes ne récidivent pas après cholécystectomie.
Le syndrome LPAC (Low Phospholipid-Associated Cholelithiasis), ou lithiase cholestérolique génétique, est une forme très particulière de lithiase biliaire qui a été décrite pour la première fois en 2001 par l’équipe de l’hôpital Saint-Antoine. Dans cette maladie, à la différence de la lithiase vésiculaire banale, il n’y a pas d’excès de sécrétion du cholestérol dans la bile mais une diminution de la sécrétion de phospholipides dont la conséquence est un défaut de formation des micelles nécessaires à la solubilisation du cholestérol, il en résulte la formation de micro-cristaux et de calculs de cholestérol (Fig. 1). Contrairement à la lithiase banale, en cas de syndrome LPAC, les calculs de cholestérol se forment aussi bien dans le foie que dans la vésicule, surviennent le plus souvent chez des patients jeunes sans surpoids et récidivent souvent après cholécystectomie [1].
Le transporteur des phospholipides (protéine MDR3, codé par le gène ABCB4) permet l’excrétion des phospholipides dans la bile au pôle canaliculaire des hépatocytes. De ce fait, des mutations du gène ABCB4 ont été logiquement identifiées comme responsables du syndrome LPAC (Fig. 1). Cependant, les mutations d’ABCB4 ne sont mises en évidence que dans un tiers à la moitié des cas de syndrome LPAC [1-3]. Des mutations d’autres gènes, non encore déterminées, sont donc probablement en cause.
Alors que le syndrome LPAC peut bénéficier d’un traitement simple et efficace par l’acide ursodésoxycholique (AUDC), la majorité des cas n’est pas diagnostiquée.
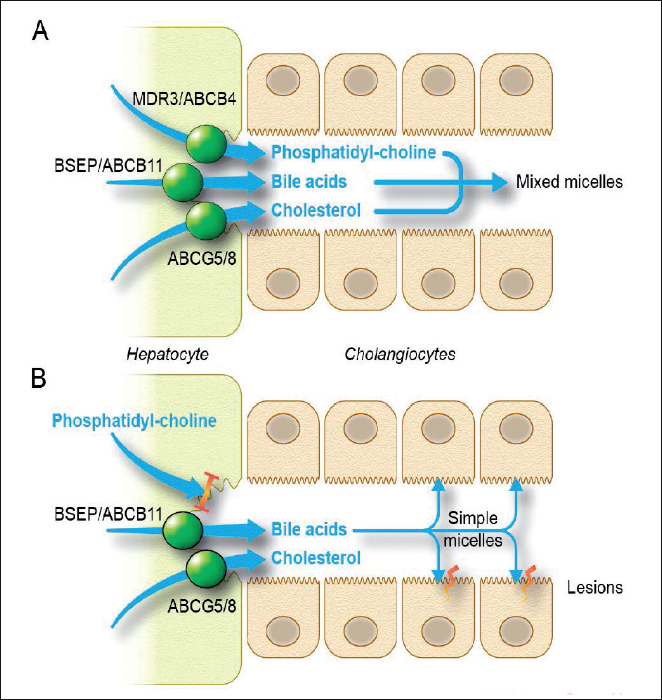
Figure 1. (A) Les transporteurs des acides biliaires, du cholestérol et de la phosphatidyl-choline (le principal phospholipide biliaire) à travers la membrane canaliculaire des hépatocytes sont normaux. Il se forme des micelles mixtes qui permettent la solubilisation du cholestérol.
(B) Quand le transporteur des phospholipides, la protéine MDR3 codée par ABCB4, est déficient, les acides biliaires sont transportés sans phospholipides et vont ainsi former de simples micelles qui n’ont pas la capacité de solubilisation du cholestérol. Il va alors se former des micro-cristaux et des calculs de cholestérol. De plus, les acides biliaires transportés sans phospholipides ont un effet détergent qui va endommager les cholangiocytes adjacents
Quand évoquer le diagnostic ? Quelles sont les caractéristiques de la maladie ?
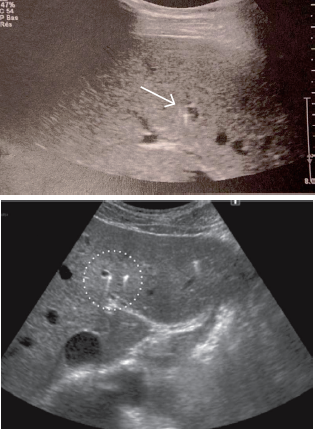
Figure 2. Spots hyper-échogène intra-hépatiques responsables d’images en queue de comète
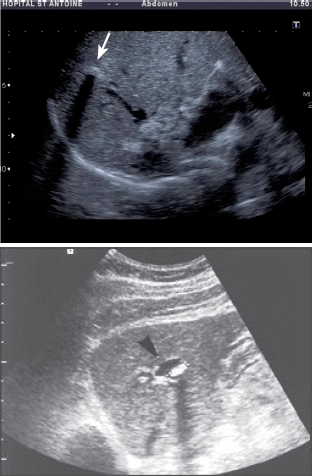
Lithiase intra-hépatique avec cône d’ombre (flèches)
En 2003, l’équipe de Saint-Antoine a comparé 18 patients porteurs de la mutation ABCB4 à des patients atteints de lithiase biliaire banale [1]. Cette équipe a ainsi pu rapporter pour la première fois que la présence d’au moins 2 des 3 critères suivants était fortement associée à la mutation ABCB4 : 1) début des symptômes avant l’âge de 40 ans ; 2) récidive des symptômes après cholécystectomie ; 3) mise en évidence par échographie de foyers hyper-échogènes intra-hépatiques avec, soit des renforcements postérieurs (« queues de comète ») correspondant à des dépôts canalaires de cristaux de cholestérol, soit des cônes d’ombre correspondant à des calculs ou micro-calculs (Fig. 2 et 3). Par ailleurs, la même équipe a montré que les caractéristiques cliniques, radiologiques et de l’analyse de la bile des patients atteints de syndrome LPAC sans mutation ABCB4 étaient identiques à celles des patients mutés pour ABCB4 [4].
Depuis 2003, les éléments permettant de distinguer le syndrome LPAC et la lithiase banale ont été précisés (Tableau I). Bien que la majorité des syndromes LPAC débute avant 40 ans, c’est l’apparition des symptômes avant 30 ans qui semble être le meilleur seuil pour distinguer le syndrome LPAC de la lithiase banale. En effet, la lithiase banale est diagnostiquée dans un tiers des cas avant 40 ans mais très rarement, en l’absence d’obésité, avant 30 ans (données personnelles de l’étude LPANGH non encore publiée qui compare 102 syndromes LPAC et 205 lithiases banales symptomatiques). La récidive des symptômes biliaires après cholécystectomie est également évocatrice de syndrome LPAC après avoir éliminé un calcul résiduel de la voie biliaire principale et la prise d’opiacés. Un antécédent personnel de cholestase gravidique est très en faveur de syndrome LPAC car ce type d’antécédent, fréquent en cas de syndrome LPAC, est exceptionnel en cas de lithiase banale. Enfin, des antécédents familiaux au premier degré de lithiase biliaire symptomatique sont retrouvés dans plus de la moitié des cas de syndrome LPAC mais ne sont pas rare en cas de lithiase banale. Ces antécédents familiaux ne sont donc évocateurs de syndrome LPAC que s’ils surviennent chez un patient jeune et non obèse.
L’âge moyen de début des symptômes du syndrome LPAC varie de 26 à 32 ans suivant les séries (étude LPANGH, 4). Les premiers symptômes de syndrome LPAC peuvent parfois survenir après 40 ans (10 % des cas suivant l’étude LPANGH) mais le début des symptômes après 50 ans est très exceptionnel. Par ailleurs, le syndrome LPAC n’est pas symptomatique avant la fin de l’adolescence ; il ne s’agit donc pas d’une cause habituelle de lithiase biliaire pédiatrique [5].
La prédominance féminine du syndrome (sex-ratio entre 1/2.3 et 1/5 suivant les études) s’explique par le rôle aggravant des œstrogènes qui inhibent la sécrétion des phospholipides dans la bile. La survenue des symptômes 10 ans plus tôt chez les femmes [4] et l’apparition fréquente des symptômes en fin de grossesse ou lors de la prise de contraceptif sont également dues au rôle aggravant des œstrogènes.
Enfin, il n’y a pas de caractéristique biologique particulière du syndrome LPAC. Une cholestase chronique a été décrite chez certains des 6 premiers patients décrits par Rosmorduc et col en 2001 [6] mais, par la suite, elle s’est avérée rare en cas de syndrome LPAC (1,4, étude LPANGH).

Tableau I. Arguments cliniques évocateurs, soit de syndrome LPAC, soit de lithiase banale en cas de lithiase biliaire symptomatique*
Comment confirmer le diagnostic ?
Devant des symptômes évocateurs, le syndrome LPAC peut être confirmé de 3 façons différentes : le génotypage ABCB4, l’analyse de la bile et l’échographie hépatique.
1) Le diagnostic génétique repose sur la mise en évidence d’une mutation significative du gène ABCB4. Si le variant du gène ABCB4 est de type non-sens le diagnostic génétique est confirmé, car ce type de mutation entraîne une altération majeure de la fonction de la protéine MDR3. Si le variant est de type faux-sens, situation la plus fréquente, son rôle pathogène devra être étayé par son référencement préalable dans des formes familiales bien documentées de syndrome LPAC ou par ses effets connus sur la fonction de la protéine MDR3. Il est donc légitime de réaliser un génotypage ABCB4 si le diagnostic de syndrome LPAC est évoqué [7]. Cependant, la recherche de la mutation ABCB4 a un coût élevé à la charge de l’établissement de santé dans lequel est fait le prélèvement. De plus, très peu de laboratoires réalisant ce génotypage, les délais d’obtention du résultat sont de plus de 1 an. Enfin, ces mutations ne sont mises en évidence que dans un tiers à la moitié des cas authentiques de syndrome LPAC. En conséquence, fait essentiel et méconnu de la prise en charge des patients atteints de syndrome LPAC, la décision d’instaurer un traitement par AUDC ne tient pas compte, dans la majorité des cas, du génotypage d’ABCB4. Il en découle que le génotypage d’ABCB4 est utile mais non indispensable pour la confirmation du diagnostic de syndrome LPAC.
2) La recherche d’un déficit en phospholipides par l’analyse de la bile [7] nécessite une expertise biochimique très spécialisée, de plus sa réalisation pratique est difficile. Elle ne peut donc pas être proposée comme élément de confirmation en pratique courante.
3) C’est la réalisation d’une échographie par un opérateur sensibilisé à la recherche des signes évocateurs qui est, en pratique, le moyen le plus pertinent pour confirmer le diagnostic. En effet, chez les patients atteints de syndrome LPAC avec mutation ABCB4, cette échographie experte révèle des anomalies typiques dans 88 à 95 % des cas (4, étude LPANGH). Cette haute pertinence diagnostique est également retrouvée en cas de syndrome LPAC sans mutation ABCB4 [4]. Le diagnostic radiologique repose sur la mise en évidence de spots hyper-échogènes intra-hépatiques responsables d’images en queue de comète de topographie compatible avec des dépôts de microcristaux le long de l’arbre biliaire (Fig. 2). Il peut aussi s’agir de sludge, d’une micro-lithiase intra-hépatique ou de macro-calculs avec cône d’ombre (Fig. 3). À l’inverse, une échographie pratiquée par un radiologue non expert et non averti du diagnostic supposé de syndrome LPAC (il s’agit le plus souvent d’échographies pratiquées en urgence devant des symptômes biliaires) ne décrira les signes de la maladie que dans 5 % des cas de syndrome LPAC (étude LPANGH).
Le plus souvent, ces anomalies radiologiques typiques de syndrome LPAC ne sont pas mises en évidence par le scanner ou l’IRM hépatique. Cependant, en complément de cette échographie experte, une bili-IRM est nécessaire pour éliminer les autres causes de lithiase intra-hépatique (essentiellement cholangite sclérosante primitive et maladie de Caroli), celle-ci étant la plupart du temps normale en cas de syndrome LPAC.
La figure 4 détaille la prise en charge diagnostique et thérapeutique du syndrome LPAC et à qui il est raisonnable de proposer une échographie par un opérateur sensibilisé.

Figure 4. Démarche diagnostique et thérapeutique du syndrome LPAC
* L’échographie hépatique réalisée par un radiologue expert est normale dans seulement 5 à 10 % des
syndromes LPAC. S’il existe une très forte suspicion clinique de syndrome LPAC (arguments nombreux
et concordants) et que l’échographie experte est normale (situation très rare), le diagnostic pourra faire
appel au génotypage d’ABCB4 et si besoin à l’analyse de la bile.
Risques évolutifs et formes particulières du syndrome LPAC
Même si certains patients ne vont rapporter que de rares épisodes de douleur biliaire, la plupart des patients non traités présente des symptômes très récidivants.
Le mode de présentation le plus fréquent du syndrome LPAC correspond à un tableau évocateur de migration lithiasique (douleur biliaire associée à une augmentation fugace des transaminases) (2, étude LPANGH). Le plus souvent, il n’y a pas de syndrome septique, donc pas d’angiocholite, et il n’est qu’assez rarement mis en évidence de calcul de la voie biliaire principale par l’écho-endoscopie ou par l’IRM biliaire. La cholécystectomie n’évite pas la récidive de ces manifestations évocatrices de migration en cas de syndrome LPAC, alors que ces symptômes disparaissent sous AUDC. De plus, un patient sur cinq atteint de syndrome LPAC présente au moins un épisode de pancréatite biliaire au cours du suivi, avant ou après cholécystectomie (étude LPANGH). La migration cholédocienne de micro-calculs d’origine intra-hépatique est donc fréquente en cas de syndrome LPAC.
À l’inverse, alors que plus de 90 % des patients atteints de syndrome LPAC présentent une lithiase vésiculaire, la cholécystite est beaucoup plus rare (aux alentours de 10 % des patients) qu’en cas de lithiase banale symptomatique (2, étude LPANGH). L’histoire naturelle du syndrome LPAC pourrait expliquer la faible incidence de la cholécystite lithiasique, la lithiase intra-hépatique étant symptomatique avant la lithiase vésiculaire avec pour conséquence une cholécystectomie souvent pratiquée à un âge jeune, prévenant ainsi le risque de cholécystite.
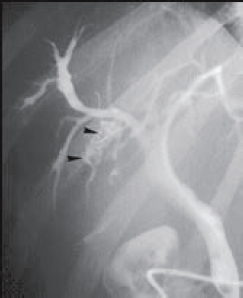
Figure 5. Dilatation sacculaire des voies biliaires intra-hépatiques autour de macro-calculs sans sténose biliaire sous-jacente (têtes de flèche)
Dans à peu près 5 % des cas, le syndrome LPAC est responsable d’une dilatation sacculaire des voies biliaires intra-hépatiques qui moulent des macro-calculs cholestéroliques sans sténose biliaire sous-jacente (Fig. 5) [8]. Le diagnostic différentiel à évoquer est la dilatation primitive congénitale des voies biliaires intra hépatiques (maladie de Caroli diffuse ou dilatation primitive localisée à un lobe ou un segment).
En cas de grossesse, la moitié des patientes atteintes de syndrome LPAC présente une cholestase gravidique [2, 4]. Ce fait n’est pas surprenant étant donné que les mutations ABCB4 sont rencontrées dans 15 % des cas de cholestase gravidique (il s’agit des mutations les plus fréquentes dans cette pathologie). La cholestase gravidique se manifeste par un prurit survenant aux 2e ou 3e trimestre associé à une cholestase biologique (diagnostiquée en cas de grossesse par une augmentation des transaminases et des acides biliaires totaux). Le pronostic est excellent pour la mère mais un accouchement prématuré est observé dans deux tiers des cas. Le traitement par AUDC à la posologie de 10 mg/kg est associé à une disparition du prurit et une diminution du risque de prématurité. À cette posologie, aucun effet indésirable n’a été rapporté. En cas de syndrome LPAC, il est donc nécessaire de prévenir les patientes de maintenir le traitement par AUDC pendant la grossesse pour éviter l’apparition d’une cholestase gravidique et son risque de prématurité mais également pour prévenir l’exacerbation des symptômes biliaires pendant la grossesse.
Enfin, malgré la publication d’exceptionnels cas de cirrhose biliaire secondaire et de cholangiocarcinome [4], et bien que le pronostic à long terme ne soit pas encore connu, le syndrome LPAC peut être considéré dans la grande majorité des cas comme étant sans gravité si le traitement par AUDC est bien suivi.
Traitement en cas de syndrome LPAC
Traitement médical
Le traitement du syndrome LPAC repose sur un traitement prolongé par AUDC à la posologie de 10 mg/kg/j (AMM obtenu en 2011 dans cette indication) [7]. L’AUDC augmente le pool des acides biliaires hydrophiles qui protègent la membrane des cholangiocytes (aux dépends des acides biliaires hydrophobes toxiques), augmente l’expression de la protéine MDR3 et facilite ainsi la sécrétion des phosholipides dans la bile [9]. Il en résulte une meilleure solubilisation du cholestérol et à distance, la dissolution des cristaux de cholestérol et des calculs. Ce traitement permet dans la majorité des cas la disparition des symptômes dès les toutes premières semaines de traitement. Cet effet est surtout spectaculaire chez les patients avec douleurs biliaires répétitives et rapprochées qui disparaissent sous AUDC. Les anomalies échographiques disparaissent beaucoup plus lentement, en quelques mois, voire en quelques années [7]. Cette disparité entre l’amélioration clinique et l’amélioration radiologique pourrait suggérer qu’une partie des symptômes serait plus liée aux cristaux de cholestérol et à l’inflammation des canaux biliaires qu’aux calculs intra-hépatiques.
L’amélioration clinique peut être partielle sous AUDC à cette posologie, avec diminution sans disparition du nombre et de l’intensité des crises douloureuses. En dehors d’une mauvaise observance du traitement par AUDC ou de douleur d’une autre origine, il peut alors être proposé d’augmenter la posologie d’AUDC jusqu’à 15 mg/kg/j. En cas d’échec de l’optimisation du traitement par AUDC, un traitement adjuvant par ezetimibe peut être proposé sur des arguments physiopathologiques, sans que la preuve de son efficacité clinique ait été apportée. En cas d’hypercholestérolémie, il est préférable d’utiliser les statines plutôt que les fibrates qui augmentent la sécrétion de cholestérol dans la bile. Enfin, le traitement œstro-progestatif doit être arrêté pendant les premières semaines de traitement par AUDC et tant que la maladie reste symptomatique ; en effet, ce traitement inhibe la sécrétion des phospholipides dans la bile et majore ainsi les symptômes. De plus, dans de rares cas, le traitement œstro-progestatif peut être responsable de cholestase en cas de syndrome LPAC.
Traitement chirurgical
Bien que la lithiase du syndrome LPAC soit fréquemment de localisation mixte (vésiculaire et intra-hépatique), les symptômes sont surtout dus à la lithiase intra-hépatique. L’efficacité du traitement médical par AUDC est telle que la cholécystectomie peut être évitée dans la majorité des cas, d’autant plus que la cholécystectomie ne prévient pas la récidive des symptômes biliaires [7]. Seuls les rares cas de cholécystite sont des indications claires à la cholécystectomie. La persistance des symptômes sous AUDC peut également faire discuter une cholécystectomie.
Dans la forme particulière de syndrome LPAC avec dilatation des voies biliaires intra-hépatiques autour de macro-calculs, le recours à la résection du segment ou du lobe atteint peut se justifier si le patient présente des angiocholites récidivantes malgré le traitement par AUDC.
Le dépistage familial
Un dépistage familial par échographie experte (et/ou génotypage si une mutation ABCB4 a été mise en évidence chez le proband) pourra être proposé aux parents du 1er degré de plus de 18 ans. Une échographie de dépistage normale chez un sujet jeune asymptomatique pourra être répétée quelques années plus tard car les signes radiologiques évocateurs de syndrome LPAC peuvent survenir ultérieurement. Le dépistage par échographie est moins rentable chez les hommes, chez qui la pénétrance des mutations ABCB4 est très incomplète. Chez les parents asymptomatiques avec lithiase intra-hépatique, il est raisonnable de proposer un traitement par AUDC.
Quelle stratégie pour ne pas passer à côté du diagnostic ?
L’étude LPANGH montre que la plupart des cas de syndrome LPAC ne sont pas diagnostiqués. En effet, dans cette étude menée dans des hôpitaux généraux de taille assez proche et disposant de services d’urgences et de chirurgie viscérale, le nombre de syndrome LPAC recensé était de 0 à 2 cas dans la plupart des hôpitaux contre 14 à 45 cas pour les 3 hôpitaux dans lesquels les équipes de chirurgie digestive, de radiologie et de gastro-entérologie sont plus particulièrement sensibilisées au diagnostic de cette maladie, sans que l’on puisse identifier de biais de sélection majeur dans ces 3 hôpitaux, les patients y étant adressés initialement pour lithiase symptomatique et non pour suspicion de syndrome LPAC.
Ce sous-diagnostic s’explique en partie par la méconnaissance d’une maladie qui n’a été décrite pour la première fois qu’il y a 15 ans. Cette méconnaissance porte sur :
- Une sous-estimation de la fréquence de la maladie, considérée généralement comme très rare, ce qui est loin d’être le cas, que ce soit à l’hôpital ou en cabinet libéral de gastro-entérologie. En effet, 20 à 25 % des femmes de moins de 30 ans présentant une lithiase biliaire symptomatique sont atteintes de syndrome LPAC [2]. Par ailleurs, à l’hôpital Saint Camille de Bry-sur-Marne (94), hôpital général de taille moyenne où sont pratiquées à peu près 200 cholécystectomies annuelles, il a été mis en évidence en moyenne, depuis 10 ans, 4 nouveaux cas de syndrome LPAC par an, soit trois à quatre fois plus que le nombre de nouveau cas de cirrhose biliaire primitive.
- La méconnaissance des signes de la maladie par les radiologues. En effet, quasiment tous les patients ont déjà eu, au moment du diagnostic, plusieurs échographies ne mettant pas en évidence les signes de syndrome LPAC, alors qu’une échographie par un radiologue expert va mettre en évidence, chez ces mêmes patients, les signes caractéristiques de LPAC dans plus de 90 % des cas.
- La méconnaissance de la maladie par les chirurgiens, qui prennent en charge la grande majorité des patients atteints de lithiase biliaire symptomatique et qui ne vont que rarement évoquer le diagnostic avant une cholécystectomie ou en cas récidive post-cholécystectomie.
La diffusion des connaissances à propos des signes cliniques qui permettent d’évoquer un syndrome LPAC auprès des collègues chirurgiens et des signes radiologiques de ce syndrome (aspect en queue de comète ou micro-lithiase intra-hépatique) auprès des collègues radiologues devrait permettre de dépister de nombreux syndrome LPAC et de transformer le cours évolutif de la maladie grâce au traitement par AUDC.
Remerciements à Marie-Pierre Hauuy (service de radiologie, hôpital Saint-Camille, Bry-sur-Marne, Val-de-Marne) et à Béatrice Noblinski (Service de radiologie, hôpital Saint-Antoine, Paris) pour l’iconographie. Remerciements à Serge Erlinger (hôpital d’Aix-en Provence, Bouches-du-Rhône) pour la figure 1. Remerciements à Christophe Corpechot (Service d’hépatologie, hôpital Saint-Antoine, Paris), à David Zanditenas (service d’hépato-gastroentérologie, hôpital Saint-Camille, Bry-sur-Marne, Val-de-Marne) et à Gilles Macaigne (service d’hépato-gastroentérologie, hôpital de Jossigny, Seine-et-Marne) pour la relecture.
Références
- Rosmorduc O, Hermelin B, Boelle PY, Parc R, Taboury J, Poupon R. ABCB4 gene mutation-associated cholelithiasis in adults. Gastroenterology 2003;125:452–59
- Condat B, Zanditenas D, Barbu V, Hauuy MP, Parfait B, El Naggar A, Collot V, Bonnet J, Ngo Y, Maftouh A, Dugué L, Balian C, Charlier A, Blazquez M, Rosmorduc O. Prevalence of low phospholipid-associated cholelithiasis in young female patients. Dig Liver Dis 2013;45:915-9.
- Pasmant E, Goussard P, Baranes L, Laurendeau I, Quentin S, Ponsot P, Consigny Y, Farges O, Condat B, Vidaud D, Vidaud M, Chen JM, Parfait B. First description of ABCB4 gene deletions in familial low phospholipid-associated cholelithiasis and oral contraceptives-induced cholestasis. Eur J Hum Genet 2012;20:277-82.
- Poupon R, Rosmorduc O, Boëlle PY, Chrétien Y, Corpechot C, Chazouillères O, Housset C, Barbu V. Genotype-phenotype relationships in the low phospholipid-associated cholelithiasis syndrome: a study of 156 consecutive patients. Hepatology 2013;58:1105-10.
- Jirsa M, Bronsky J, Dvorakova L, Sperl J, Smajstrla V, Horak J, Nevoral J, Hrebícek M. ABCB4 mutations underlie hormonal cholestasis but not pediatric idiopathic gallstones. World J Gastroenterol 2014;20:5867-74.
- Rosmorduc O, Poupon R, Hermelin B. MDR3 gene defect in adults with symptomatic intrahepatic and gallbladder cholesterol cholelithiasis. Gastroenterology 2001:1459–1467.
- Rosmorduc O, Poupon R. Low phospholipid-associated cholelithiasis: association with mutation in the MDR3/ABCB4 gene. Orphanet J Rare Dis 2007;2:29.
- Poupon R, Arrive L, Rosmorduc O. The cholangiographic features of severe forms of ABCB4/MDR3 deficiency-associated cholangiopathy in adults. Gastroenterol Clin Biol 2010;34:380-7.
- Erlinger S. Low phospholipid-associated cholestasis and cholelithiasis. Clin Res Hepatol Gastroenterol 2012;36:S36-S40.
Les Cinq points forts
- Les patients atteints de syndrome LPAC ont souvent des épisodes de douleurs biliaires multiples et rapprochés et/ou un tableau évocateur de migration cholédocienne accompagnée parfois de pancréatite ; en revanche la cholécystite est rare.
- Les arguments évocateurs de syndrome LPAC sont : le début des symptômes avant 30 ans, la récidive des symptômes après cholécystectomie, les antécédents personnels de cholestase gravidique, les antécédents familiaux au premier degré de lithiase biliaire symptomatique avant 30 ans.
- Des mutations du gêne ABCB4 sont mises en évidence dans un tiers à la moitié des cas de syndrome LPAC.
- La réalisation d’une échographie par un opérateur sensibilisé à la recherche des signes du syndrome LPAC (queues de comètes intra-hépatiques) est le moyen le plus pertinent pour confirmer le diagnostic.
- Le traitement est l’AUDC. La cholécystectomie doit être réservée aux rares cholécystites.
FMC HGE : Organisme certifié Qualiopi pour la catégorie ACTIONS DE FORMATION