La cirrhose biliaire primitive : actualités
Objectifs pédagogiques :
- Quels sont les éléments du diagnostic de cirrhose biliaire primitive et la place de la ponction biopsie hépatique ?
- Modalités du traitement, surveillance et causes d’échec.
- Prise en charge des formes avec réponse partielle.
- Place de la transplantation hépatique.
Généralités
La cirrhose biliaire primitive (CBP) est une maladie chronique du foie caractérisée sur le plan clinique par un syndrome de cholestase intra-hépatique et sur le plan anatomopathologique par une destruction progressive des petits canaux biliaires1. En l’absence de traitement, la maladie évolue vers un état fibreux du foie pouvant conduire à l’hypertension portale (HTP), à l’insuffisance hépatique, puis au décès. La fréquence des anticorps anti-mitochondries (AAM) et la forte prédominance féminine font de cette maladie l’archétype de la maladie auto-immune du foie. La CBP n’a pas d’étiologie connue. Les études familiales, génétiques et épidémiologiques suggèrent qu’elle résulte d’une combinaison complexe de facteurs de risque héréditaires et environnementaux. La CBP est ubiquitaire et atteint selon les pays entre 20 et 400 individus par million d’habitants.2 Sa prévalence est largement sous-estimée du fait de la fréquence des formes mineures asymptomatiques.Rapportée à la population à risque, la maladie toucherait environ une femme de plus de 40 ans surmille. La CBP évolue schématiquement en 3 phases : une phase asymptomatique pouvant durer plus de 10 ans et passer totalement inaperçue en l’absence d’une analyse systématique des tests biologiques hépatiques; une phase symptomatique marquée par une fatigue, un prurit,une cholestase biologique progressive et le développement d’une fibrose hépatique et d’une ductopénie; enfin, une phase terminale caractérisée par un ictère permanent associé à une altération de l’état général, à une cirrhose, à une HTP, puis à une insuffisance hépatique conduisant au décès en 2 à 4 ans si une transplantation hépatique (TH) n’est pas envisageable. Des formes ductopéniques rapides ou des formes frontières associées à une hépatite auto-immune (HAI, syndrome de chevauchement) sont plus rares. Le diagnostic précoce est important car l’efficacité du traitement médical dépend en grande partie du stade auquel il est débuté. La prise au long cours d’acide ursodésoxycholique (AUDC) permet de ralentir la maladie et d’augmenter l’espérance de vie des patients mais ce traitement reste insuffisant chez 30% à 40%d’entre eux. A ce jour, il n’y a pas d’autre médicament dont l’efficacité sur la survie ait été établie. La TH, lorsqu’elle est possible, reste le traitement de choix des formes terminales de la maladie.
Quels sont les éléments du diagnostic de cirrhose biliaire primitive et la place de la ponction biopsie hépatique ?
Le diagnostic de CBP doit être évoqué devant toute élévation chronique inexpliquée de l’activité sérique des tests hépatiqueschez une femme de plus de 35 ans, surtoutsi elle prédomine surles phosphatases alcalines (PAL) ou la gamma-glutamyltranspeptidase (GGT).Le sexe masculin n’exclue pas le diagnostic (10% des cas) mais le rend moins plausible. L’existence d’un prurit isolé sans lésion cutanée préalable est un bon signe d’appel mais il n’est présent que dans 30 à 50% des cas. Son absence n’exclue donc pas le diagnostic. La fatigue est le principal symptôme allégué par les patients mais elle est totalement non spécifique. Des douleurs articulaires, un syndrome sec buccal ou oculaire, un syndrome de Raynaud,unxanthelasmaou une hépatomégalie peuvent aussi êtreprésents.Les formes historiquement décrites dans les années 60 révélées d’emblée par un ictère et une altération de l’état général sont devenues très rares.
Selon les recommandations 2009 de l’American Association for the Study of LiverDiseases (AASLD),3 le diagnostic de CBP peut être établi si au moins 2 des critères suivants sont présents : 1) signes biochimiques de cholestase reposant principalement sur l’activité sérique des PAL ; 2) présence d’AAM ; 3) lésion de cholangite destructrice non suppurée des canaux biliaires interlobulaires.Publiées la même année, les recommandations de l’European Association for the Study of the Liver (EASL)4indiquent que le diagnostic de CBP peut être formellement porté chez un adulte ayantune élévation inexpliquée des PAL et une recherched’AAM positive (la biopsie n’étant réservée dans cette situation qu’à l’évaluation du stade de la maladie).A noter qu’il n’existe pas de consensus concernantle seuil critique d’activité des enzymes hépatiques pour le diagnostic de CBP.L’AASLD ne donne pas d’indication. L’EASL définit la cholestase biologique parune activité des PAL >1,5 fois la limite supérieure de la normale (N) associée une GGT >3 x N.A noter également que l’augmentation de la concentration sérique des IgM,bien que fréquemment observée, ne fait officiellement pas partie des critères diagnostiques retenus pas l’AASLD et l’EASL.
Les AAM sont présents chez 90-95% des patients atteints de CBP. Ils sont recherchés en première intention par immuno-fluorescence indirecte (IFI) sur lame triple substrat (foie, rein et estomac de rat)5. Un titre ≥ 1/40 chez des patients ayant des signes de cholestase peut être considéré comme significatif.6 Cette positivité en IFI doit toujours être confirmée par un test spécifique de reconnaissance d’un ou de plusieurs antigènesmitochondriaux recombinants qui peut différer selon les laboratoires (ELISA, dot blot, western blot). Des anticorps anti-nucléaires (AAN) sont présents chez 25% à 70% des patients atteints de CBP.Deux types d’AAN ont une forte spécificité (> 90%) pour la CBP : 1) des AAN avec fluorescence membranaire cerclée (« rim-like »), dirigés contre les pores nucléaires et correspondant dans 50% des cas à des anti-gp210 lorsqu’ils sont spécifiquement recherchés par ELISA, 2) des AAN avec fluorescence en « gros grains »(« dot blot ») intranucléairescorrespondant à des anti-sp100 dirigés contre les cores nucléaires.7 Ces AAN ont une faible sensibilité (≤ 25%) pour le diagnostic de CBP mais peuvent êtreprésents dans près de 50% des cas de CBP sans AAM. A la différence des AAM, ils pourraient avoir une valeur pronostique car ils semblent être plus fréquents dans les formes sévères et évolutives de la maladie. Des anticorps anti-muscle lisse (AML) sont rarement observés en dehors des cas d’HAI associée (syndrome de chevauchement).A noter que des AAM sont parfoisdétectés en l’absence de tout signe biologique ou histologique de CBP.La séroprévalence des AAM dans la population générale a été évaluée à0,5%des donneurs de sang lorsqu’un test sensible (ELISA) est utilisé. Dans cette situation, il faut rechercher une hépatite virale C, une autre maladie autoimmune(en particulier HAI, sclérodermie, syndrome des anti-phospholipides,anémie hémolytique auto-immune),unedysglobulinémie et une prise de papavérine. Les patients ayant des AAM isolés sans anomalies des tests enzymatiques hépatiques sont à risque de développer une CBP et doivent avoir un dosage des PAL une fois par an.
La ponction-biopsie hépatique (PBH) n’est pasnécessaire au diagnosticdans la majorité des cas.En revanche, elledemeureindispensable en cas de forme atypique, c’est à dire d’absence d’AAM (CBP « séronégative ») oud’augmentation inhabituelle des transaminases (> 5 xN) ou des IgGsériques (> 20 g/L)pouvant suggérerun syndrome de chevauchement (HAI), ou de toute autre pathologie hépatique suspectée.3La PBH garde toute sa placedans l’évaluation de la sévérité de la maladie et en particulier du degré d’activité inflammatoire, de la fibrose et de la paucité biliaire. La taille de la PBH est essentielle. La probabilité d’observerdeslésionsbiliaires augmente avec le nombre d’espaces porte. L’examen de 10 à 15 espaces porte et de plusieurs plans de coupe est nécessaire à l’évaluation qualitative et quantitative des canaux biliaires. La lésion caractéristique de la CBP est la cholangite destructrice granulomateusenon suppurée (dite lésion biliaire« floride »).La cholangite destructrice lymphocytaire non granulomateuse est une variante moins spécifique. Les lésions de cholangite destructrice non suppuréesont très évocatrices de CBP sans être totalement pathognomoniques (Tableau 1). Il est important de noter que ces lésions peuvent être absentes de l’échantillon examinésans pour autant exclure le diagnostic car leur répartition est hétérogène et segmentaire le long des canaux interlobulaires. D’autres signes non spécifiques ont une valeur d’orientationdiagnostique vers une maladie biliaire: inflammation portale prédominante, granulome épithélioïde, cholestase histologique périportale, réaction ductulaire, ductopénie.La ductopénie est habituellement définie par un pourcentage d’espaces porte sans canaux biliaires supérieur à 50%.3 Un immuno-marquage des canaux biliaires par la cytokératine 7peut faciliterson évaluation. La présence d’une activité nécrotico-inflammatoire marquée, périportale ou lobulaire, doit suggérer l’existence d’un syndrome de chevauchement (HAI) mais il n’est pas rare d’observer une hépatite d’interface lymphocytaire segmentaire et focale au cours de la CBP.Le stade histologique tient compte à la fois du degré d’inflammation et de fibrose (classifications de Ludwig8et deScheuer9). Le stade de Scheuerprend égalementen compte la présence ou non de lésions biliaires floridesou d’une réaction ductulaire. Quelque soit la classification, le stade est définien 4 catégories(Figure 1).C’est le niveau lésionnel le plus avancé sur la biopsie qui définit le stade de la maladie: atteinte portale (stade 1), atteinte périportale (stade 2), atteinte septale (stade 3), cirrhose (stade 4).
L’élastométriehépatique (Fibroscan)est une méthode concurrentielle d’évaluationdu stade de la CBP. Elle a d’excellentes valeurs prédictives positive et négative pour la détection ou l’exclusiond’une fibrose sévère (≥F3) ou d’une cirrhose10. Elle est moins performante pour différencier les stades intermédiaires de fibrose (≤F2). Elle pourrait éviter la réalisation de la PBH dans le seul but d’affirmer ou d’éliminerl’existence d’une fibrose extensive.Toutefois, il n’y a aucune recommandation officielle d’utilisation de l’élastométrie dans le bilan initial de la CBP, l’AASLD se contentant d’évoquer son intérêt potentiel3. Plusieurs tests sériques de fibrose ont été évalués au cours de la CBP (acide hyaluronique,scores de Forns, APRI,FIB-4 et ELF).Mais ces marqueurs n’ont pas fait l’objet d’étude extensive de sorte que leur utilisation ne peut être recommandée en pratique clinique.
Ilest important de noter qu’il n’existe pas de critères consensuels pour le diagnostic de syndrome de chevauchement entre la CBP et l’HAI.Du fait des implications thérapeutiques potentielles, il est cependant recommandé d’éliminer ce diagnostic dès qu’un diagnostic de CBP a été établi. Lesscores diagnostiques de l’HAI dans leur version originale, révisée11 ou simplifiée12ne sont pas adaptés. Les critères diagnostiques de Paris13(Tableau 2) basés sur la présence d’au moins 2 critères d’HAIont été recommandés par l’EASL.4
Modalités du traitement, surveillance et causes d’échec.
Le traitement de fond de la CBP repose sur l’administration orale d’AUDCau long cours à la dose de 13 à 15 mg/kg/j. Ce traitement a un effet bénéfique bien établi sur les taux de bilirubine, des enzymes hépatiques (en particulier les PAL), de cholestérol et des IgM.14 L’effet thérapeutique sur la progression de la maladie etla survie en particulier a été plus difficile à établir en raison de l’hétérogénéité des essais cliniques, du faible nombre de patients et des durées de suivi insuffisantes.L’analyse des données combinées des 3 premiers essais effectués contre placebo (français, canadien, américain)15a montré que la survie sans TH des patients traités par AUDC pendant 4 ans était supérieure à celle des patients ayant reçu soit un placebo pendant 4 ans soit un placebo pendant 2anssuivi d’un traitement en « ouvert » par AUDC pendant encore 2 ans(Figure 2).L’essai de la Mayo Clinic montrait en outre une diminution de l’incidence des varices oesophagiennes sous AUDC. Ces résultats ont été cependant réfutéspar la méta-analysede l’ensemble des essais thérapeutiques publiés n’ayant pas retrouvé d’effet significatif du traitement sur le risque de décès, deTH ou de complications de la maladie.16 Les résultats de cetteméta-analyse sont critiquables car plus de la moitié des essais inclus étaient de durée inférieure à 2 ans ouévaluaient des posologiesnon optimales d’AUDC.Une seconde méta-analyse effectuée à partir de l’ensemble des essais ayant testé l’AUDC à une posologie ≥ 11 mg/kg/j pendant une période d’au moins 2 ans a confirmé l’existence d’un bénéfice thérapeutique sur la survie sans TH ainsi qu’une diminutionde laprogression histologique à partir des stades précoces17. Enfin, l’effet de l’AUDC sur le prurit et la fatigue est discordant selon les essais et ne ressortpas de manière significativedans les méta-analyses.
Les recommandations de l’EASL et de l’AASLD sont de traiter par AUDC (13-15 mg/kg/j) de manière prolongée tous les patients atteints de CBP quelque soit leur stade, même si ils sont asymptomatiques.3, 4 L’AUDC est administré le plus souvent en 2 (parfois 3) prises orales. En cas d’administration parallèle d’un chélateur des acides biliaires pour le traitement du prurit (cholestyramine, colestipol)les prises d’AUDC doivent être espacées d’au moins 2 à 4 heures par rapport à la prise du chélateur. En cas de maladie sévère ou très fortement symptomatique (cirrhose, HTP, ictère, prurit intense)il est recommandé de débuter l’AUDC à faible dose (200-250 mg/j)et d’augmenter de 200-250 mgtoutes les semaines jusqu’à la dose optimalesous surveillance du taux de bilirubine.L’AUDC est en principe contre-indiqué au cours du 1er trimestre de la grossesse et au cours de l’allaitement, mais aucune toxicité pour le fœtus ou le nouveau-né n’a été incidemment rapportéechez les patientes ayant conçuet mené leur grossesse et l’allaitement sous AUDC. L’AUDC a peu d’effets secondaires. Une diarrhée transitoire est observée en début de traitement chez une minorité de patients(< 5%). Des brûlures épigastriquespeuvent être rapportées de façon épisodique.Une prisede poids de 1 à 2 kg est statistiquementobservéela première année.La réponse thérapeutique à l’AUDC doit être évaluée à l’aide des tests biochimiques hépatiques mesurés tous les 3 à 6 mois selon la sévérité initiale de la maladie.L’amélioration des PAL et de la GGT s’observe dès le premier mois et90%de la réponse biochimique est atteintà1 an. Vingt pourcent des patients ont une normalisation complète des tests hépatiques après 2 ans de traitement.
Les traitements non spécifiques concernent la prise en charge du prurit, la prévention ou le traitement de l’ostéoporose, l’hypercholestérolémie et l’HTP.Le traitement du prurit doit faire appel en 1ère intentionà la cholestyramine (4-16 g/j) qui doit être prise à distance de l’AUDC (2-4 h),puis en cas d’échec à la rifampicine (150-600 mg/j)qui est souvent plus efficace, enfin si nécessaireà la naltrexone (25-50 mg/j)ou à la sertraline(25-100 mg/j)d’efficacitévariableet souvent limités par leurs effets secondaires. Les anti-histaminiquesbien que non recommandésde manière spécifique en raison de leur effet sédatif sont assez largement prescritsdans la pratique.La photothérapie par UVB pourrait avoir un effet bénéfique mais il n’existe pas d’étude de grande taille. Dans les cas de prurit réfractaireil est nécessaire de recourir à des traitements invasifscomme les échanges plasmatiques, le système MARS (épuration extracorporelle sur colonne de charbon et gradient d’albumine) ou le drainagenaso-biliaire avant d’envisager la TH en cas d’insuffisance hépatique associée.Concernant le risque d’ostéoporose, il est recommandé de suppléer tous les patients en calcium (1 g/j) et vitamine D (800 UI/jou 100000 UI/3-4 mois) même si le bénéfice n’est pas formellement démontré. Un traitement par alendronate (70 mg/semaine) est indiquéen cas d’ostéoporose (T score < 2,5) ou d’antécédent de fracture porotique. En cas de varices oesophagiennes, une perfusion mensuelle de biphosphonatedoit être préférée.L’hypercholestérolémie au cours de la CBP n’est pas associée à une augmentation démontrée du risque cardio-vasculaire et l’intérêt d’un traitement hypocholestérolémiantdans cette situation est discutable.Toutefois, en cas d’hypercholestérolémie familiale ou de maladie cardiovasculaire avérée une statinepeut être prescritesans risque particulier pour le foie.Enfin, le traitement de l’HTP n’est pas différent de celui recommandé dans les autres causes d’HTP intra-hépatique.
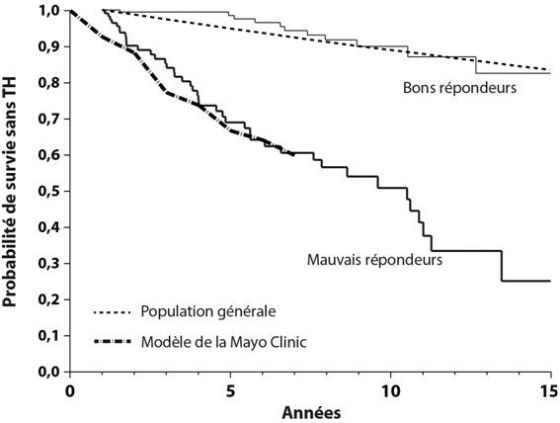
La survie des patients traités par AUDC reste inférieure à celle d’une population témoin appariée pour l’âge et le sexe.18 Les principaux facteurs de mauvais pronostic sont : 1) en début de traitement : une bilirubine totale élevée (> 17 µmoles/L), un stade histologique ≥ 3, une mesure de la dureté du foie > 9,6 kPaet l’existence sur la PBH d’une hépatite d’interface significative (segmentaire et focale dans la majorité des espaces porte ou circonférentielle dans plus de 50% d’entre eux); 2) sous traitement : une réponse biochimique à 1 an insatisfaisante et une augmentation de la dureté du foie > 2,1 kPa/an.Les indicateurs pronostiques non spécifiques (dont les scores de Child-Pugh et MELD) sont valides au stade de cirrhose. La réponse biochimique à 1 an est l’un des facteurs les plus fortement liés au pronostic.Il existe plusieurs définitions de la réponse biochimique incomplète (Tableau 3).19-23. Les critères de Paris, qui ont été validés dans différentes populations, sont recommandés par l’EASL et l’AASLDdans le butd’identifier les patients mauvais répondeurs à l’AUDC.3, 4 . Ces patients ont une survie sans TH de 51%à 10 ans alors qu’elle est de 90% chez les répondeurs (Figure 3).21L’augmentation de la dureté du foie mesurée parélastométrie est probablement un bon critère de réponse incomplète à l’AUDC.10 Cependant,il n’existe pas d’étude prospective démontrantde façon formelle l’intérêt de l’élastométriedans le suivi des patients et aucune recommandation officielle n’a été faite à ce sujet.
Prise en charge des formes avec réponse partielle.
En cas de réponse incomplète à l’AUDC, il est recommandé derechercher de manière systématique les situations suivantescar elles peuvent déboucher surdes mesures thérapeutiques spécifiques efficaces: 1) posologie d’AUDC insuffisante ; 2) observance sous-optimale; 3) syndrome de chevauchement (HAI) ; 4)dysthyroïdie ; 5) maladie coeliaque.Il est bien établi qu’une dose d’AUDC ≤ 10 mg/kg/j est associée à une moins bonne réponse biochimique.24Un dosage chromatographique ou spectrométrique desacides biliaires sériques primaires, secondaires et tertiairesn’est pas effectué en pratique courante car c’est un examen couteux, non totalementstandardisé et réservé à un nombre de laboratoirestrès restreint.Toutefois, en cas de mauvaise réponse thérapeutique, cet examenpourrait s’avérerutile.En effet, le pourcentage d’AUDC circulant est assez bien corrélé à son enrichissement biliaire qui est lui même bien corrélé à la baisse des PAL.Unpourcentage d’AUDC circulant< 50% est considéré comme insuffisant, la fourchette thérapeutique étant comprise entre 50% et 80%. La recherche de signes d’HAI en cas de mauvaise réponse à l’AUDC (voir critères diagnostiquesdu syndrome de chevauchement au chapitre précédent) est primordiale car les formes mixtes de CBPsont observées dans 10% des caset répondentle plus souvent à un traitement complémentairepar corticoïdes plus ou moins associés à l’azathioprine.Les dysfonctionnements thyroïdiens doivent être recherchés car ils sontfréquentsau cours de la CBP (10%-15%) etpourraientparticiper aux anomalies de la biologiehépatique. La maladie coeliaquequi est retrouvée chez < 1% des patients atteints de CBP peut être une caused’anomalies persistantes des tests hépatiques sous AUDC.
Dans la majorité des casaucun de ces facteurs n’est retrouvé et la questionposée est de savoir si il est nécessaire d’optimiser le traitement par AUDC et si oui, comment?Pour le moment,il n’existe pas de réponse évidente à ces questions. L’AASLD reconnaît que des essais thérapeutiquessont justifiés chez lespatients ayant une réponse biochimique incomplète après 3 à 6 mois d’AUDC.25 Les doses élevées d’AUDC (≥ 20 mg/kg/j) n’améliore pas la réponse biochimique24. L’addition de colchicine, de methotrexateou de mycophenolatemofetiln’apporte pasde bénéfice thérapeutique. Les traitements semblant pouvoir améliorer la réponse à l’AUDCsont les corticostéroïdes, en particulier le budésonide, les fibrates (agonistes du récepteurnucléaire PPARα) et les dérivés synthétiques de l’acide chenodésoxycholique dont l’acide obeticholique (agonistes du récepteur nucléaire des acides biliaires). L’adjonction de prednisone (ou prednisolone) à la dose de 10 à 30 mg/j, associée ou non à l’azathioprine, améliore les tests biochimiques et l’inflammation histologique aux stades précoces de la maladie mais augmente significativement le risque d’ostéopénie.Le budésonide à la dose de 6 à 9 mg/jsemble avoir les mêmes effets bénéfiques mais avec un risque d’ostéopénie cortico-induite moins élevé26, 27 .Cependant son bénéfice sur la progression cliniqueet la surviereste à démontrer (un essai thérapeutique est en cours). A noter qu’il est contre-indiqué chez les patients cirrhotiques en raison d’un passage systémique accru augmentant le risque de complications métaboliques et osseuseset d’un risque rapporté de thrombose de la veine porte.Les fibrates (fenofibrate, bezafibrate) en association à l’AUDC ou en monothérapie ont un effet bénéfique certain sur les tests biochimiques hépatiques28mais il n’y a pas pour le moment d’essai thérapeutique prolongé prouvant leur intérêt sur le long terme (plusieurs essais sont en cours). L’acide obeticholique (AOC) est un agoniste du récepteur des acides biliaires FXR (Farnesoid X receptor) dont le rôle protecteur au cours de la cholestase est bien établiin vitro et in vivo. Cette molécule en cours d’évaluation constitue la 1ère génération d’agonistes synthétiques de FXR.Desessais de courte durée ont montré qu’elle avait un effet bénéfique sur les tests biochimiques hépatiques en cas de réponse incomplète à l’AUDC, mais au prix d’une augmentation de la fréquence et de l’intensité du prurit pour des doses > 10 mg/j.29 Là encore, le bénéfice en termes de survie sans complication reste à démontrer (un essai prolongé est en cours).
Place de la transplantation hépatique.
La TH a considérablement amélioré le pronostic des formes terminales de la maladie. La survieà 5ans après TH est de 80%-85%. Les indications de la TH ne diffèrent pas de celles des autres causes d’insuffisance hépatique et les règles établies sur le score MELD ou de Child-Pughrestent valides.La concentration de la bilirubine totale a néanmoins une valeur indicative spécifique. Les patients dont la bilirubineapproche 100 µmol/L (~6 mg/dL) doivent être orientés vers un centre de TH.30 Le score de la Mayo Clinic, qui tient compte deplusieursparamètres pronostiques (âge, bilirubine, albumine, temps de Quick,présence d’oedème ou d’une ascite)peut également participer à la décision (scorecritique ≥ 7,8) mais son calcul peu aisé fait qu’il est rarement utilisé en pratique clinique.Les patients souffrant d’une forme ductopénique rapide se manifestant par un ictère et un prurit sévèrenon réversibles doivent être orientés vers un centre de TH même en l’absence de signe clinique ou histologique de cirrhose.Un prurit réfractaire isolé, c’est à diresans ictère ou autre signe de gravité,n’est pas considéré comme une indication en soi à la TH.Le risque de récidive clinique de la maladie est estimé à 20-25% après 10 ans. Ce risque serait supérieur en cas de traitement partacrolimus(vs. ciclosporine) mais le niveau de preuve n’est pas assez élevé pour donner lieu à des recommandations spécifiques. L’arrêt des corticoïdes n’est pas conseillé car le risque de rejet est élevé.Les récidives sur le greffon sont généralement peu sévère et répondent souvent de manière favorableà la réintroduction de l’AUDC. Les cas dere-transplantations pour récidivesévère de la maladie sur le greffonsont rares.
Tableau 1. Causes de cholangite destructrice non suppuréedes canaux interlobulaires
Cirrhose biliaire primitive
Cholangite sclérosante primitive ou secondaire
Sarcoïdose
Hépatite auto-immune
Cholangite médicamenteuse
Cholangite néoplasique (lymphome, mastocytose, hystiocytose)
Hépatite C, B, E
Réaction du greffon contre l’hôte (allogreffe de moelle)
Tableau 2. Critères de Paris pour le diagnostic d’HAI quand le diagnostic de CBP a été établi (au moins 2 critères doivent être présents)
1) Alanine aminotransférase (ALAT) > 5 x N
2) IgG> 2 x N ou AML ≥ 1/80
3) Hépatite d’interface périportale ou périseptale modérée ou sévère
Tableau 3. Critères de réponse biochimique incomplète à l’AUDC.
| Désignation | Durée de traitement |
Critères |
| Rochester | 6 mois | PAL ≥ 2 x N ouscore de la Mayo ≥ 4.5 |
| Barcelone | 12mois | Diminution desPAL ≤ 40% |
| Paris (I) | 12mois | PAL ≥ 3 x N ou AST ≥ 2 x N ouBili. Tot.>17 µmole/L |
| Paris (II) * | 12mois | PAL ≥ 1,5x N ou AST ≥ 1,5x NouBil. Tot.>17 µmole/L |
| Rotterdam | 12mois | Bilirubine Totaleet/ou Albumine anormale |
| Toronto** | 24mois | PAL > 1.67 x N |
| Ehime | 6 mois | Diminutionde la GGT ≤ 70% |
* spécifiques des stades histologiques précoces (1-2). ** défini sur la base d’une progression histologique d’au moins 1 stade (tous les autres critères ont été définis sur la base de la survie sans TH).
Figure 1. Les 4 stades histologiques de la CBP selon la classification de Scheuer : A, Stade 1 avec lésion de cholangite destructrice granulomateuse (flèche) ; B, Stade 2 avec réaction ductulairepériportale (flèche) ; C, Stade 3 avec fibrose septale extensive sans cirrhose; D, stade 4 correspondant à la cirrhose (A – B, HES x100 ; C, rouge sirius x50 ; D, rouge sirius x20).

Figure 2. Taux de survie sans transplantation hépatique évalués à partir de 548 patients traités initialement par AUDC (13-15 mg/kg/j) ou placebo (analyse combinée de 3 grands essais thérapeutiques)

Figure 3. Taux de survie sans transplantation des patients traités par AUDC en fonction de la réponse biochimique à 1 an (critères de Paris).
Remerciements :
L’auteur remercie le Pr Dominique Wendum (service d’Anatomie Pathologique, Hôpital Saint-Antoine, Paris) qui a gentiment accepté de fournir les illustrations histologiques.
Références
- 1. Selmi C, Bowlus CL, Gershwin ME, Coppel RL. Primary biliary cirrhosis. Lancet 2011;377:1600-9.
- 2. Gross RG, Odin JA. Recent advances in the epidemiology of primary biliary cirrhosis. Clin Liver Dis 2008;12:289-303; viii.
- 3. Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ. Primary biliary cirrhosis. Hepatology 2009;50:291-308.
- 4. EASL EAftSotL. EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol 2009;51:237-67.
- 5. Invernizzi P, Lleo A, Podda M. Interpreting serological tests in diagnosing autoimmune liver diseases. Semin Liver Dis 2007;27:161-72.
- 6. Vergani D, Alvarez F, Bianchi FB, Cancado EL, Mackay IR, Manns MP, Nishioka M, Penner E, International Autoimmune Hepatitis G. Liver autoimmune serology: a consensus statement from the committee for autoimmune serology of the International Autoimmune Hepatitis Group. J Hepatol 2004;41:677-83.
- 7. Invernizzi P, Selmi C, Ranftler C, Podda M, Wesierska-Gadek J. Antinuclear antibodies in primary biliary cirrhosis. Semin Liver Dis 2005;25:298-310.
- 8. Ludwig J, Dickson ER, McDonald GS. Staging of chronic nonsuppurative destructive cholangitis (syndrome of primary biliary cirrhosis). Virchows Arch A Pathol Anat Histol 1978;379:103-12.
- 9. Scheuer P. Primary biliary cirrhosis. Proc R Soc Med 1967;60:1257-60.
- 10. Corpechot C, Carrat F, Poujol-Robert A, Gaouar F, Wendum D, Chazouilleres O, Poupon R. Noninvasive elastography-based assessment of liver fibrosis progression and prognosis in primary biliary cirrhosis. Hepatology 2012;56:198-208.
- 11. Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, Chapman RW, Cooksley WG, Czaja AJ, Desmet VJ, Donaldson PT, Eddleston AL, Fainboim L, Heathcote J, Homberg JC, Hoofnagle JH, Kakumu S, Krawitt EL, Mackay IR, MacSween RN, Maddrey WC, Manns MP, McFarlane IG, Meyer zum Buschenfelde KH, Zeniya M, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol 1999;31:929-38.
- 12. Hennes EM, Zeniya M, Czaja AJ, Pares A, Dalekos GN, Krawitt EL, Bittencourt PL, Porta G, Boberg KM, Hofer H, Bianchi FB, Shibata M, Schramm C, Eisenmann de Torres B, Galle PR, McFarlane I, Dienes HP, Lohse AW, International Autoimmune Hepatitis G. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008;48:169-76.
- 13. Chazouilleres O, Wendum D, Serfaty L, Montembault S, Rosmorduc O, Poupon R. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome: clinical features and response to therapy. Hepatology 1998;28:296-301.
- 14. Lindor K. Ursodeoxycholic acid for the treatment of primary biliary cirrhosis. N Engl J Med 2007;357:1524-9.
- 15. Poupon RE, Lindor KD, Cauch-Dudek K, Dickson ER, Poupon R, Heathcote EJ. Combined analysis of randomized controlled trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology 1997;113:884-90.
- 16. Gong Y, Huang Z, Christensen E, Gluud C. Ursodeoxycholic Acid for Patients With Primary Biliary Cirrhosis: An Updated Systematic Review and Meta-Analysis of Randomized Clinical Trials Using Bayesian Approach as Sensitivity Analyses. Am J Gastroenterol 2007;102:1-9.
- 17. Shi J, Wu C, Lin Y, Chen YX, Zhu L, Xie WF. Long-term effects of mid-dose ursodeoxycholic acid in primary biliary cirrhosis: a meta-analysis of randomized controlled trials. Am J Gastroenterol 2006;101:1529-38.
- 18. Poupon RE, Bonnand AM, Chretien Y, Poupon R. Ten-year survival in ursodeoxycholic acid-treated patients with primary biliary cirrhosis. Hepatology 1999;29:1668-71.
- 19. Angulo P, Lindor KD, Therneau TM, Jorgensen RA, Malinchoc M, Kamath PS, Dickson ER. Utilization of the Mayo risk score in patients with primary biliary cirrhosis receiving ursodeoxycholic acid. Liver 1999;19:115-21.
- 20. Pares A, Caballeria L, Rodes J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic Acid. Gastroenterology 2006;130:715-20.
- 21. Corpechot C, Abenavoli L, Rabahi N, Chretien Y, Andreani T, Johanet C, Chazouilleres O, Poupon R. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology 2008;48:871-7.
- 22. Kuiper EM, Hansen BE, de Vries RA, den Ouden-Muller JW, van Ditzhuijsen TJ, Haagsma EB, Houben MH, Witteman BJ, van Erpecum KJ, van Buuren HR. Improved prognosis of patients with primary biliary cirrhosis that have a biochemical response to ursodeoxycholic acid. Gastroenterology 2009;136:1281-7.
- 23. Azemoto N, Kumagi T, Abe M, Konishi I, Matsuura B, Hiasa Y, Onji M. Biochemical response to ursodeoxycholic acid predicts long-term outcome in Japanese patients with primary biliary cirrhosis. Hepatol Res 2011;41:310-7.
- 24. Angulo P, Dickson ER, Therneau TM, Jorgensen RA, Smith C, DeSotel CK, Lange SM, Anderson ML, Mahoney DW, Lindor KD. Comparison of three doses of ursodeoxycholic acid in the treatment of primary biliary cirrhosis: a randomized trial. J Hepatol 1999;30:830-5.
- 25. Silveira MG, Brunt EM, Heathcote J, Gores GJ, Lindor KD, Mayo MJ. American Association for the Study of Liver Diseases endpoints conference: design and endpoints for clinical trials in primary biliary cirrhosis. Hepatology 2010;52:349-59.
- 26. Leuschner M, Maier KP, Schlichting J, Strahl S, Herrmann G, Dahm HH, Ackermann H, Happ J, Leuschner U. Oral budesonide and ursodeoxycholic acid for treatment of primary biliary cirrhosis: results of a prospective double-blind trial. Gastroenterology 1999;117:918-25.
- 27. Rautiainen H, Karkkainen P, Karvonen AL, Nurmi H, Pikkarainen P, Nuutinen H, Farkkila M. Budesonide combined with UDCA to improve liver histology in primary biliary cirrhosis: A three-year randomized trial. Hepatology 2005;41:747-752.
- 28. Itakura J, Izumi N, Nishimura Y, Inoue K, Ueda K, Nakanishi H, Tsuchiya K, Hamano K, Asahina Y, Kurosaki M, Uchihara M, Miyake S. Prospective randomized crossover trial of combination therapy with bezafibrate and UDCA for primary biliary cirrhosis. Hepatol Res 2004;29:216-222.
- 29. Mason A, Luketic VA, Lindor K, Hirschfield GM, Gordon SC, Mayo MJ, Kowdley KV, Pares A, Trauner M, Sciacca CI, Beecher Jones T, Pruzanski M, Shapiro DA. Farnesoid – X Receptor Agonists: a New Class of Drugs for the Treatment of PBC? An International Study Evaluating the Addition of Obeticholic Acid ( INT – 747 ) to Ursodeoxycholic Acid. Hepatology 2010;52:357A.
- 30. Neuberger J. Liver transplantation for primary biliary cirrhosis: indications and risk of recurrence. J Hepatol 2003;39:142-8.
LES POINTS FORTS MR CORPECHOT
- Le diagnostic de cirrhose biliaire primitive ne nécessite pas de PBH dans 90% des cas.
- Un syndrome de chevauchement (cirrhose biliaire primitive -hépatite auto-immune) est présent dans 10% des cas.
- Le traitement par acide ursodésoxycholique est bien toléré et efficace notamment aux stades précoces.
- La survie sans transplantation hépatique est corrélée à la réponse biochimique sous acide ursodésoxycholique
- Il n’existe pas d’autre traitement validé excepté la transplantation hépatique au stade terminal.
FMC HGE : Organisme certifié Qualiopi pour la catégorie ACTIONS DE FORMATION
FMC HGE : Organisme certifié Qualiopi pour la catégorie ACTIONS DE FORMATION