Biomarqueurs des cancers colorectaux utiles en pratique clinique
Objectifs pédagogiques
- Connaître les biomarqueurs utilisés en clinique dans le diagnostic et la prise en charge des CCR
- Connaître l’impact diagnostique, pronostique et théranostique des biomarqueurs
- Savoir quand, sur quel matériel et à qui demander la recherche des biomarqueurs du CCR
- Savoir interpréter les résultats en fonction de la situation clinique
On définit sous le nom de biomarqueurs du cancer colorectal (CCR), les marqueurs biologiques qui influencent sa prise en charge thérapeutique. Ceux-ci peuvent être sériques (ex : ACE, cellules tumorales circulantes, ADN circulant), tumoraux (ex : IMS, RAS, BRAF) ou constitutionnels (ex : DPD, UGT1A1, FcgRIIA). Nous n’aborderons ici que les marqueurs tumoraux, ceux-ci tenant une place prédominante au sein des biomarqueurs du CCR utiles en pratique clinique à l’heure actuelle.
Ces biomarqueurs sont issus d’une meilleure compréhension des mécanismes moléculaires impliqués dans la cancérogénèse colorectale. En effet, bien que les CCR soient des tumeurs homogènes sur le plan anatomopathologique, les progrès récents de la biologie moléculaire ont permis d’individualiser différents mécanismes moléculaires de carcinogenèse colorectale. On distingue aujourd’hui deux principaux mécanismes de cancérogenèse colorectale [1] :
- l’instabililité chromosomique (CIN : Chromosomal INstability ou LOH : Loss Of Heterozygoty) impliquée dans environ 80 % des CCR ;
- l’instabilité microsatellitaire (IMS) impliquée dans 12 à 15 % des CCR.
Plus récemment, un troisième mécanisme a été identifié : l’instabilité épigénétique, associé à une hyperméthylation de l’ADN au niveau de certaines régions promotrices de plusieurs gènes (CIMP : CpG Island Methylation Phenotype) induisant une inactivation transcriptionnelle de gènes suppresseurs de tumeur. Ce 3e groupe n’est cependant pas totalement autonome des 2 autres.
Malgré cette hétérogénéité moléculaire, les conséquences en termes d’altération des voies de signalisation sont communes. Les principales voies de signalisation impliquées sont :
- la voie RAS/RAF/MAPKinase,
- la voie PI3K/AKT.
Nous présentons les principaux biomarqueurs tumoraux en décrivant leurs valeurs pronostiques et prédictives et leurs applications diagnostiques et/ou thérapeutiques en pratique clinique aujourd’hui. On entend par valeur pronostique, l’impact du biomarqueur indépendamment de tout traitement (histoire naturelle de la maladie) alors que la valeur prédictive concerne l’impact du biomarqueur sur l’efficacité d’une thérapeutique donnée.
Les différents biomarqueurs tumoraux des CCR
Instabilité microsatellitaire (IMS)
L’IMS (ou MSI pour MicroSatellite Instability) caractérise l’une des 2 voies majeures de carcinogenèse (Fig. 1).

Figure 1. Les 2 principales voies de carcinogenèse colorectale. a) voie CIN (ou LOH) ; b) voie MSI. D’après Vilar E, et al. Nat Rev Clin Oncol 2010 March;7(3):153-62.
L’autre voie, la plus fréquemment impliquée (environ 80 % des CCR), est la voie de l’instabilité chromosomique (CIN ou LOH). Elle est caractérisée par des pertes alléliques sur le bras court des chromosomes 17 et 8 et sur le bras long des chromosomes 18, 5 et 22. Ces pertes alléliques sont associées à des mutations fréquentes des gènes suppresseurs de tumeurs (TP53, APC, SMAD4…) et participent ainsi à l’inactivation bi-allélique de ces gènes.
La voie de l’IMS est caractérisée par la présence d’une instabilité des « microsatellites ». Les microsatellites sont des séquences répétées de l’ADN particulièrement exposées à des erreurs à type de mésappariements lors de la réplication de l’ADN. Normalement ces erreurs sont réparées par un complexe de protéines appartenant au système MMR (Mismatch Repair) comprenant les protéines MLH1, MSH2, MSH6 et PMS2. L’IMS est liée à un défaut de réparation des mésappariements de l’ADN survenus au cours de la réplication. Ce défaut de réparation est lié à une inactivation bi-allélique de gène du système MMR (gènes MLH1, MSH2, MSH6, PMS2). Cette inactivation est le plus souvent due à une hyperméthylation de la région promotrice du gène MLH1. Cette déficience du système de réparation induit une accumulation de mutations secondaires au sein de gènes impliqués dans le contrôle tumoral (TGFßRII, BAX, TCF-4…).
La voie IMS est impliquée dans la genèse de 12 à 15 % des CCR sporadiques [2]. Le phénotype tumoral est alors appelé IMS ou dMMR (deficient MisMatch Repair), en opposition aux tumeurs à microsatellites stables (MSS).
Ce mécanisme de carcinogenèse est aussi en cause dans les tumeurs développées dans le cadre d’un syndrome de Lynch défini par l’existence d’une mutation constitutionnelle d’un gène MMR. Quasiment toutes les tumeurs développées dans le cadre d’un syndrome de Lynch présentent une IMS.
Facteur pronostique de l’IMS
Le facteur pronostique des CCR de phénotype IMS est clairement établi pour les CCR de stade localisé : dès 2003 le travail de Ribic a montré qu’au sein des CCR de stade II et III, ceux de phénotype IMS étaient de meilleur pronostic que les MSS [2]. Ces résultats ont ensuite été confirmés par de nombreuses études ou méta-analyses [3, 4].
Cet impact pronostique bénéfique semble concerner l’ensemble des stades. En effet, les études s’adressant aux CCR tous stades confondus (incluant des stades métastatiques) affichent en effet une réduction nette du risque de décès [5, 6].
Plus récemment, il est apparu que la coexistence d’une mutation du gène BRAF (mutation V600E, retrouvée dans 40 à 50 % des cas de CCR sporadique avec IMS) doit être prise en compte. En effet, deux études montrent clairement qu’au sein des CCR IMS de stade II-III seuls ceux sans mutation V600E BRAF sont de bon pronostic, alors que ceux avec mutation V600E BRAF ont un pronostic intermédiaire [7, 8].
Facteur prédictif de l’IMS
L’IMS peut avoir un impact sur l’efficacité de la chimiothérapie adjuvante des CCR de stade localisé. Deux situations doivent être distinguées : celle de la chimiothérapie adjuvante par 5FU seule et celle de la chimiothérapie adjuvante par FOLFOX.
Chimiothérapie adjuvante par 5FU seul
Les CCR de stade II-III de phénotype IMS ne semblent tirer aucun bénéfice de survie d’une chimiothérapie adjuvante par 5FU seul, contrairement aux CCR MSS. Cette donnée initialement rapportée par l’étude de Ribic a été clairement retrouvée, à la fois en terme de survie sans récidive et de survie globale, dans plusieurs études, analyses poolées et méta-analyse [2, 3].
Il a même été suggéré que la chimiothérapie adjuvante par 5FU seul des CCR IMS de stade II pouvait non seulement être inefficace mais, de surcroît, délétère… [2] et que l’un des mécanismes de résistance au 5FU résiderait dans l’expression de la thymidylate synthase qui serait plus élevée au sein des tumeurs IMS que MSS [10]. Ces données restent à confirmer.
Enfin, il est aussi possible que la valeur prédictive de l’IMS sur l’inefficacité de la chimiothérapie adjuvante par 5FU dépende du mécanisme d’IMS : elle concernerait les CCR IMS sporadiques (par hyperméthylation MLH1) et ne concernerait pas les CCR IMS dans le cadre d’un syndrome de Lynch. Les données cliniques restent cependant limitées et doivent être confirmées [11].
Chimiothérapie adjuvante par FOLFOX
La valeur prédictive péjorative du statut IMS semble limitée à la chimiothérapie adjuvante par 5FU seul et ne pas concerner la chimiothérapie par FOLFOX. En effet, d’une part il n’existait pas d’interaction entre le statut IMS et le bénéfice de l’oxaliplatine au cours de l’étude NSABP-C07 évaluant FUFOL ± Oxaliplatine [12] et d’autre part l’analyse du sous-groupe de patients IMS de l’étude MOSAIC montre un maintien du bénéfice du FOLFOX par rapport au LV2FU2 (Flejou JF, ASCO 2013, abst 3524). Ces données restent cependant limitées par le faible effectif étudié et mériteront d’être confirmées.
Conséquences cliniques
Le statut IMS a donc un impact dans le domaine des CCR localisés et notamment des CCR de stade II pour lesquels se discute l’indication d’une chimiothérapie adjuvante, c’est-à-dire les stades II dits « à haut risque ». Le statut IMS conférant un très bon pronostic (au moins pour les tumeurs sans mutation BRAF), il n’est pas recommandé de réaliser une chimiothérapie adjuvante pour les CCR IMS.
Ainsi, le TNCD précise clairement que « La détermination du statut microsatellite stable (MSS) ou instable (IMS) de la tumeur est devenue indispensable pour poser l’indication d’une chimiothérapie adjuvante pour un patient opéré d’un cancer de stade II avec facteurs de mauvais pronostic ».
Méthodes de détermination de l’IMS
Il y a deux méthodes possibles pour analyser le statut IMS d’une tumeur. Les deux méthodes sont à la fois complémentaires et très bien corrélées :
- par biologie moléculaire (PCR) : la recherche d’une IMS se pratique sur ADN tumoral en étudiant 5 séquences microsatellitaires mononucléotidiques quasi-monomorphes. La présence d’une IMS se traduira par une différence de taille des séquences microsatellitaires de l’ADN tumoral par rapport à l’ADN normal ;
- par immuno-histochimie sur coupes en paraffine : il s’agit alors de rechercher la perte d’expression de protéines du système MMR. Une perte d’expression tumorale de protéine MMR traduit une altération génétique tumorale du gène MMR correspondant. Il s’agit donc d’un test reflétant le même défaut MMR que celui responsable de l’IMS visualisée par biologie moléculaire.
Voies de signalisation
Voie de l’EGF (Epidermal Growth Factor)
Le récepteur de l’EGF (EGF-R), ou HER1, est une protéine transmembranaire de la classe des récepteurs à activité tyrosine kinase et de la famille des récepteurs HER (HER1, HER2 ou ErbB2, HER3 et HER4). Il est composé d’un domaine extracellulaire assurant la fixation du ligand, d’un domaine transmembranaire et d’un domaine effecteur tyrosine-kinase intracellulaire (Fig. 2).
 Figure 2. Récepteur EGF-R
Figure 2. Récepteur EGF-R
Il existe plusieurs ligands, dont l’EGF, le TGFa, l’amphiréguline et l’épiréguline. La fixation du ligand entraîne l’activation du récepteur, après sa dimérisation, par autophosphorylation de résidus tyrosine spécifiques situés dans son domaine intracellulaire. Cette phosphorylation déclenche l’activation de deux voies de signalisation intracellulaire d’aval : la voie RAS / RAF / MEK / MAPK et la voie PI3K / AKT / m-Tor. Ces voies activent, in fine, la transcription de gènes impliqués dans la prolifération, la migration, l’adhésion et la différenciation cellulaire, ainsi que dans la résistance à l’apoptose et l’angiogenèse (Fig. 2).
Une sur-activation de la voie de l’EGF est impliquée dans un grand nombre de CCR et le blocage de cette voie est devenu une arme thérapeutique ciblée efficace. Plusieurs mécanismes peuvent expliquer cette sur-activation : essentiellement une augmentation de l’expression des récepteurs à la surface de la cellule (30 à 80 % des cas) et/ou une augmentation de la quantité de ligand, et certainement beaucoup moins -fréquemment au cours des CCR que d’autres types de cancer, une activation constitutive du récepteur ou des voies RAS / RAF / MEK / MAPK ou PI3K / AKT.
L’efficacité thérapeutique du blocage de la voie EGF a mené, depuis 2004, à l’autorisation de mise sur le marché, dans la prise en charge des CCR, de deux anticorps monoclonaux ciblant l’EGF-R : le cetuximab (IgG1 chimérique) et le panitumumab (IgG2 humain). Tous deux agissent comme des antagonistes dirigés contre le domaine extracellulaire de liaison de l’EGF-R, en compétition avec le ligand. Ces progrès thérapeutiques ont participé à l’allongement net de la médiane de survie des patients métastatiques non résécables, actuellement estimée entre 24 et 34 mois.
La prescription des anticorps anti-EGFR avait été initialement restreinte aux patients dont la tumeur exprimait l’EGFR en immunohistochimie. Toutefois, l’expression de la protéine ainsi mesurée n’était pas corrélée à l’obtention d’une réponse ou à la survie ; ce paramètre a donc été abandonné en 2008. De même, ni l’évaluation de l’amplification du gène de l’EGF-R par hybridation in situ, ni celle de l’expression de ligand tel que l’amphi- et l’épiréguline n’ont pu trouver de place en tant que biomarqueurs en pratique clinique [13]. En revanche, à partir de 2006 il est apparu qu’un facteur clé de la réponse aux anti-EGFR était le statut mutationnel des gènes codant pour les protéines RAS. Cette découverte a mené à des avancées remarquables en terme de ciblage des thérapies anti-EGFR.
Voie RAS
Les protéines RAS font partie de la famille des GTPases. Il en existe 4 isoformes codées par trois gènes différents : KRAS (Kirsten RAS), HRAS (Harvey RAS) et NRAS (Neuroblastoma RAS). Elles jouent un rôle important dans la transmission, vers le noyau, de signaux extracellulaires provenant de récepteurs membranaires et notamment de l’EGF-R.
RAS sont des protéines cytosoliques qui nécessitent leur translocation sur la face interne de la membrane cellulaire pour être activées. Leur activation est déclenchée majoritairement par les récepteurs membranaires à tyrosine kinase dont l’EGF-R. Une fois activées elles activent à leur tour les voies de signalisation d’aval aboutissant aux phénomènes de prolifération, invasion, migration, etc.
RAS joue un rôle « d’interrupteur » et oscille entre un état actif et un état inactif. Sous sa forme active, RAS recrute et active les 2 principales voies de signalisation de l’EGFR : celle des MAP kinases (RAS / RAF / MEK / MAPK) par le biais de la protéine RAF et celle des PI3K / AKT (cette dernière voie pouvant aussi être activée par l’EGFR sans l’intermédiaire de RAS) (Fig. 3).
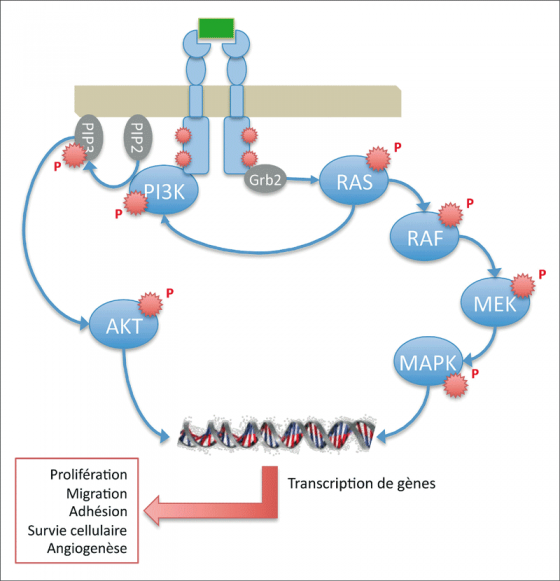
Figure 3. Voies RAS/RAF/MAK/MAPK et PI3K/AKT
Dans les CCR, le gène KRAS est fréquemment muté (40 à 50 % des cas) ; la plupart des mutations (40 %) se trouvent dans l’exon 2 du gène KRAS, le plus souvent au niveau des codons 12 et 13. Dans moins de 10 % des cas, elles concernent l’exon 3 (codons 61 et 59) et l’exon 4 (codons 117 et 146). Le gène NRAS est beaucoup plus rarement porteur de mutations (environ 5 à 8 % des cas). Enfin, il n’est pas décrit de mutation du gène HRAS au cours des CCR. Au total environ 50 à 60 % des CCR présentent une mutation RAS.
Ces mutations, sont des mutations faux sens qui aboutissent à une forme de RAS bloquée en phase active. Constamment activée, elle active elle-même les voies de signalisation d’aval qui stimulent la prolifération, survie, différenciation, migration cellulaire et angiogenèse… [14]
Facteur pronostique
Il ne semble pas que le statut mutationnel RAS ait une valeur pronostique. Cependant la question fait encore débat, les résultats d’études étant contradictoires tant dans les formes localisées que dans les formes métastatiques.
Facteur prédictif
À partir de 2006, des premiers travaux ont montré que la présence d’une mutation des codons 12 et 13 de l’exon 2 du gène KRAS était associée à une résistance au cetuximab [15, 16]. Ces résultats ont ensuite été validés par plusieurs études portant sur des patients traités par cetuximab et panitumumab dans le cadre d’essais contrôlés [13]. Ainsi, depuis 2008 et compte tenu de l’ensemble de ces travaux, l’obtention de l’AMM des anti-EGFR a été conditionnée par le génotypage KRAS, limitant l’indication aux CCR sans mutation KRAS des codons 12 et 13 de l’exon 2.
En 2013, le ciblage thérapeutique des anti-EGFR dans le CCR métastatique a connu une nouvelle avancée significative avec la validation de l’impact prédictif d’autres mutations situées sur les gènes KRAS et NRAS. En effet, des travaux récents réalisés chez des patients chimio-réfractaires traités par panitumumab seul et en première ligne traités par FOLFOX + panitumumab (étude PRIME) ou FOLFIRI + cetuximab (étude FIRE-3) ont permis de démontrer que la présence d’autres mutations KRAS (codons 61, 117 et 146) retrouvées chez environ 10 % des patients et de mutations du gène NRAS (codons 12, 13, 61, 117 et 146), retrouvées chez environ 6 %, étaient également prédictives de la non efficacité du traitement (17, Heinemann J, ESMO 2013 abst. 17). Ces résultats ont débouché sur une modification de l’AMM restreignant l’indication aux CCR dits « RAS sauvages » c’est-à-dire sans mutation KRAS ni NRAS sur l’ensemble des codons précités.
Ce meilleur ciblage, en excluant plus de 50 % des CCR clairement résistants aux anti-EGFR, permet d’améliorer les résultats thérapeutiques de cette classe médicamenteuse dans le sous-groupe de patients sans facteur de résistance identifié. Il permet aussi de limiter les effets directement délétères d’un tel traitement. En effet, l’étude PRIME nous confirme que l’adjonction de panitumumab au FOLFOX chez les patients porteurs de CCR RAS muté, non seulement n’améliore pas les résultats par rapport au FOLFOX seul mais, de surcroît, est délétère à la fois en terme de survie sans progression (10,1 mois vs 7,9 mois, HR = 0,72) et de survie globale (26 mois vs 20,2 mois, HR = 0,78) [17]. De même, l’étude FIRE-3 suggère que l’association FOLFIRI-cetuximab est délétère, par rapport au FOLFIRI-bevacizumab, chez les patients avec mutation RAS (SSP : 6,1 vs 12,2 mois, p = 0,004) (Heinemann J, ESMO 2013 abst. 17).
Conséquences cliniques
Les AMM des deux thérapies anti-EGFR (panitumumab et cetuximab) disponibles dans le traitement des CCR métastatiques ont été modifiées, respectivement, en juillet et décembre 2013. Il est précisé que la détermination du statut mutationnel RAS de type sauvage (exons 2, 3 et 4 du KRAS et du NRAS) est impérative avant l’instauration d’un tel traitement et qu’il doit être déterminé par un laboratoire expérimenté utilisant une méthode d’analyse validée.
Cependant, si la présence d’une mutation RAS est liée à une résistance au traitement par anti-EGFR voire à un effet délétère et contre-indique un tel traitement, l’absence de mutation RAS ne garantit pas une réponse aux anti-EGFR (30 à 40 % des patients restent non répondeurs). Ainsi, d’autres mécanismes de résistance restent à identifier, potentiellement dans les étapes d’aval de la voie de signalisation.
BRAF
Les protéines RAF sont des protéines sérine/thréonine – Kinases. Il en existe 3 isoformes codées par trois gènes différents : A-RAF, B-RAF et C-RAF.
La protéine BRAF appartient à la voie de signalisation RAS / RAF / MEK / MAPK mise en jeu lors de l’activation de l’EGF-R. Son rôle est bien établi dans la carcinogenèse colorectale : située « en aval » de RAS. La protéine BRAF est activée par RAS et elle va activer à son tour MEK, elle-même activant ERK qui induit, après translocation, l’expression de gènes impliqués dans la prolifération et l’apoptose (Fig. 3).
Les mutations du gène BRAF sont retrouvées dans 5 à 10 % des CCR. Dans plus de 95 % des cas, il s’agit d’une mutation à type de transversion T > A (remplacement d’une thymine en une adénine) appelée communément mutation BRAF V600E. Cette mutation est activatrice : elle mène à une forme activée de la protéine BRAF. Elle induit donc une activation de la voie RAS / MAPK similaire à l’activation provoquée par les mutations de gènes RAS.
La mutation BRAF V600E est mutuellement exclusive de mutations du gène RAS ; ceci pourrait s’expliquer par l’implication de chacune d’elle comme mutation « driver » activatrice de la même voie de signalisation. Elle est plus fréquente dans les tumeurs de localisation proximale (25 à 30 %) que distale (moins de 5 %) et survient beaucoup plus fréquemment dans les cancers sporadiques IMS que dans les tumeurs MSS (respectivement 50-80 % et 5-10 %) [18].
Facteur pronostique
La valeur pronostique de la mutation BRAF V600E dans les CCR métastatiques est bien établie au travers de nombreuses études : elle confère à la tumeur un très mauvais pronostic, et ce quels que soient les traitements reçus. Sa présence est associée à une nette diminution de la survie globale (au moins deux fois plus de risque de décès), particulièrement pour les tumeurs MSS [18, 19].
Son rôle pronostique reste plus discuté pour les formes localisées de CCR, les résultats des études ne sont pas concordants mais il pourrait être aussi un facteur de mauvais pronostic induisant une diminution de la survie sans récidive. Il apparaît avoir un impact pronostique au moins au sein du sous-groupe des CCR localisés de phénotype IMS : sa valeur pronostique péjorative impacte la valeur pronostique favorable de IMS (cf. chapitre « Facteur pronostique de l’IMS ») [8, 9].
Facteur prédictif
Le rôle prédictif de résistance aux anti-EGFR de mutations BRAF dans le traitement des CCR est largement suspecté par un rationnel préclinique fort et des analyses rétrospectives de séries cliniques non randomisées et non contrôlées [20]. Néanmoins, ces données n’ont pas pu être clairement confirmées par les analyses issues des essais de phase III ayant évalué les anti-EGFR en situation métastatique, les résultats de ces différentes analyses étant discordants. Une des raisons pourrait être liée aux faibles effectifs de patients mutés au sein des études. De plus, l’impact pronostique péjoratif de cette mutation est tel qu’il « écrase » les résultats et pourrait masquer un éventuel impact prédictif. En effet, avant d’avoir une valeur prédictive potentielle de la réponse aux Ac anti-EGFR, les mutations BRAF ont surtout une valeur pronostique péjorative dans le CCR métastatique.
En pratique à l’heure actuelle, il n’y a donc pas de contre-indication de prescription des anti-EGF-R en présence d’une mutation BRAF.
Thérapies ciblées anti-BRAF
Les thérapies spécifiquement ciblées anti-BRAF (vemurafenib, dabrafenib…), utilisées en monothérapie au cours d’études de phase précoce, ont donné jusqu’à présent des résultats assez décevants dans le traitement des CCR avec mutation BRAF par rapport à ces mêmes thérapies utilisées pour le traitement des mélanomes BRAF mutés (plus de 50 % d’entre eux) où elles ont obtenu l’AMM. De même le sorafenib, thérapie multicible incluant BRAF (et anti-angiogène), a donné des résultats décevants en phase II.
Les études doivent néanmoins se poursuivre, notamment en association avec les inhibiteurs de MEK, d’EGFR ou d’AKT au vu de données précliniques et d’études de phase I encourageantes.
Conséquences cliniques
À ce jour, il n’y a pas de recommandation de prise en charge spécifique des patients porteurs CCR métastatiques avec mutation BRAF. Notamment la prescription d’Ac anti-EGFR n’est pas contre-indiquée, contrairement aux CCR avec mutations RAS. Cependant, la connaissance de ce statut tumoral peut influencer la prise en charge en incitant à un traitement de chimiothérapie intensifiée dès la 1re ligne, compte tenu de l’agressivité de la maladie et peut-être à privilégier une trithérapie de type FOLFOXIRI [21]. De plus, elle incite à adresser précocement les patients vers des essais évaluant des thérapies ciblées anti-BRAF.
Syndrome de Lynch ; conséquences diagnostiques
La très faible incidence de mutation BRAF dans les CCR IMS liés à un syndrome de Lynch (1,4 %) par rapport aux CCR sporadiques (50 à 80 %) permet d’utiliser cette caractéristique comme aide diagnostique au syndrome de Lynch.
En effet, dans le cadre de la recherche d’un syndrome de Lynch, il est recommandé d’analyser le statut IMS pour tout CCR diagnostiqué à moins de 60 ans ou, quel que soit l’âge, en cas d’antécédent personnel ou familial au 1er degré de CCR ou autre cancer du spectre HNPCC [22]. En l’absence de statut IMS, le diagnostic de syndrome de Lynch peut être écarté (le phénotype IMS étant quasiment constant dans le contexte de syndrome de Lynch). En présence de statut IMS le diagnostic de syndrome de Lynch reste compatible ; deux situations distinctes doivent être envisagées :
- Première situation, s’il existe une perte d’expression tumorale de la protéine MSH2 et/ou MSH6 en immunohistochimie et un maintien de l’expression de MLH1 : ce profil est alors caractéristique d’un syndrome de Lynch et doit mener à une prise en charge oncogénétique spécifique avec recherche de mutation constitutionnelle des gènes MSH2 et MSH6.
- Deuxième situation, plus fréquente, s’il existe une perte d’expression tumorale de la protéine MLH1 : ce profil est compatible avec un syndrome de Lynch mais il pourrait aussi s’agir d’un CCR sporadique de statut IMS (par hyperméthylation somatique du promoteur MLH1). L’identification d’une mutation BRAF V600E oriente vers le diagnostic de CCR sporadique et permet de surseoir à l’enquête oncogénétique approfondie et à la recherche de mutation constitutionnelle (sauf si la présentation clinique et/ou familiale est évocatrice de syndrome de Lynch). En l’absence de mutation BRAF V600E, la question du syndrome de Lynch reste posée. Dans ce dernier cas, la recherche d’hyper-méthylation tumorale du promoteur de MLH1 peut apporter une aide en orientant vers une origine sporadique ; dans les autres cas, une recherche de mutation constitutionnelle du gène MLH1 doit être menée.
L’émergence de nouveaux biomarqueurs
Il est probable que d’autres biomarqueurs soient utilisés en pratique clinique à court terme. Quelques pistes sont largement ouvertes :
- La voie PI3K / AKT, activée entre autres par EGF-R, pourrait trouver sa place dans la prédiction de l’efficacité de l’aspirine dans le traitement adjuvant des CCR localisés. En effet, l’aspirine permettrait de diminuer considérablement le risque de rechute des CCR avec mutation de PIK3CA (gène codant pour la sous-unité catalytique de la PI3K) soit 15 à 20 % des CCR [23].
- C-MET est un récepteur tyrosine kinase dont le gène est amplifié dans 10 à 20 % des CCR. Il pourrait être un biomarqueur de mauvais pronostic et de résistance secondaire aux anti EGF-R. Il constitue une cible thérapeutique en cours d’évaluation dans le CCR avancé. L’étude de son expression immunohistochimique au niveau tumoral ou de l’amplification de son gène pourrait ainsi devenir un outil de pratique clinique demain.
Comment faire une recherche de biomarqueurs tumoraux du CCR en pratique
Biologie moléculaire
L’ensemble des biomarqueurs précités se détermine par technique de biologie moléculaire (recherche d’IMS ou recherche de mutation de gènes).
L’analyse des biomarqueurs est effectuée sur un fragment de tumeur, le plus souvent fixé et inclus en paraffine et conservé dans un laboratoire d’anatomie-pathologique. En France, les analyses de biomarqueurs à visée diagnostiques, théranostiques ou pronostiques sont réalisées dans une des 28 plateformes hospitalières de génétique moléculaire des cancers mise en place par l’INCa et réparties sur le territoire français. La liste de ces différentes plateformes, les coordonnées des laboratoires effectuant les analyses et les référents pour chaque type de biomarqueurs sont disponibles sur le site de l’INCa (www.inca.fr).
Cependant, les plateformes ne sont pas détentrices du matériel tumoral et la demande d’analyse de biomarqueurs doit donc être adressée à l’anatomo-pathologiste détenteur du matériel tumoral dont les coordonnées sont généralement inscrites sur les comptes-rendus d’anatomo-pathologie. Il faut utiliser pour cela des fiches de prescription spécifiques qui mentionnent le type de recherche prescrite (mutations RAS, test IMS, mutation BRAF…). Ces fiches sont proposées par les plateformes de génétique ou bien par les laboratoires pharmaceutiques. Elles ont l’avantage d’être standardisées et permettent de renseigner les éléments indispensables au bon déroulement de l’analyse. Outre l’identité du patient, la date et le type de prélèvement, il est aussi indispensable de renseigner correctement les coordonnées du clinicien prescripteur afin de lui adresser le résultat. Dès réception de cette demande et après avoir choisi le bloc tumoral le plus adapté pour l’analyse, le pathologiste doit transmettre dans les plus brefs délais le matériel tumoral à la plateforme, accompagné de la fiche de prescription qu’il aura complétée.
Le circuit de l’analyse est donc le suivant :
1) prescription de l’analyse moléculaire par le clinicien et envoi de cette prescription à l’anatomo-pathologiste détenteur du matériel tumoral,
2) transmission par le pathologiste de la fiche de prescription accompagnée du matériel tumoral à la plateforme de génétique des tumeurs,
3) réalisation de l’analyse sur la plateforme et renvoi du résultat au clinicien prescripteur et à l’anatomo-pathologiste (avec le bloc tumoral).
Sur quel type de matériel peut-on réaliser une analyse moléculaire ?
Biopsie ou résection chirurgicale ? Tumeur primitive ou métastase ? Le seul facteur limitant est la quantité de cellules tumorales dans le prélèvement qui peut être parfois très limité (biopsie ou cancer du rectum réséqué après radio-chimiothérapie). C’est le pathologiste qui vérifie s’il existe suffisamment de cellules tumorales dans le matériel conservé avant de l’envoyer à la plateforme pour l’analyse. En cas de matériel tumoral non disponible (zone tumorale épuisée ou trop petite pour une analyse moléculaire), il devra en informer le clinicien prescripteur afin de rechercher un autre prélèvement ou de programmer une autre biopsie.
Les analyses sont généralement réalisées sur du matériel tumoral fixé et inclus en paraffine. Il faut utiliser un fixateur adéquat pour la biologie moléculaire et éviter les fixateurs à base d’acide. Le fixateur de choix est le formol tamponné neutre. Le temps de fixation peut également altérer l’ADN. La fixation des biopsies dans le formol ne doit donc pas dépasser 24 heures pour les biopsies et 48 heures pour les résections chirurgicales. Afin de maîtriser les temps de fixation, le médecin préleveur (endoscopiste, radiologue, chirurgien) doit donc prendre l’habitude de noter l’heure de fixation du prélèvement pour toute demande d’examen anatomo-pathologique.
Quelles sont les étapes de l’analyse moléculaire ?
Les anomalies recherchées dans le CCR sont des mutations ponctuelles (RAS, BRAF) ou bien des variations de taille et de nombre d’allèles pour un locus donné (IMS). Ces anomalies sont donc recherchées sur de l’ADN extrait à partir d’un petit fragment de tumeur, après une étape d’amplification de l’ADN (phase pré-analytique). Le choix de la zone tumorale à extraire s’effectue sous contrôle microscopique, par un pathologiste, pour un enrichissement maximal en cellules tumorales.
Les techniques d’analyse ensuite utilisées (phase analytique) sont variables d’une plateforme à l’autre mais sont toutes basées sur de l’amplification d’ADN. Pour la recherche de mutations, la technique classique de séquençage Sanger est de plus en plus remplacée par des techniques plus sensibles, soit de séquençage amélioré « pyroséquençage » ou dérivé « Snapshot » ou bien par des techniques dites ciblées car seul le brin d’ADN muté sera amplifié (amplification allèle spécifique, discrimination allélique, etc.). La sensibilité de ces techniques est de l’ordre de 5 % (détection de 5 % de mutants dans un échantillon).
La recherche d’IMS est beaucoup plus standardisée et repose sur la recherche de variation de taille d’au moins 5 marqueurs microsatellitaires dits monomorphes après amplification des loci correspondants ; une tumeur étant instable quand au moins 2 des 5 marqueurs testés sont instables. La détection d’une mutation (ou d’instabilité microsatellitaire) dans un échantillon dépend donc de trois paramètres principaux : la qualité de l’ADN extrait (taille de la portion d’ADN que l’on peut amplifier, < 150 pb généralement suffisant pour la recherche de mutations, mais > 150 pb pour la recherche d’instabilité microsatellitaire), de la proportion de cellules tumorales dans l’échantillon extrait et la sensibilité de la technique. Il est admis qu’en deçà de 20 % de cellules tumorales dans l’échantillon extrait, il existe un risque de faux-négatif.
Le délai d’une analyse moléculaire simple sur la plateforme est de l’ordre de 8 à 10 jours, délai auquel il faut ajouter le temps d’acheminement du bloc tumoral vers la plateforme et le délai de transmission du résultat vers le clinicien prescripteur. La réalisation de ces analyses moléculaires nécessite donc une bonne coordination entre les cliniciens, les pathologistes et les plate-formes de génétique.
Vers d’autres techniques ?
À l’heure actuelle, on recherche les mutations une à une (technique dite « unitaire ») et chacune d’entre elles sont réalisées successivement en utilisant une certaine quantité d’ADN. Plus le nombre de mutations à rechercher augmente (ex : 6 exons différents pour RAS), plus le délai d’analyse augmente et plus on consomme de l’ADN. L’année 2014 va voir le développement, sur les plateformes, de techniques dites de « haut-débit » (ex : NGS ou séquenceur de nouvelle génération) qui permettront l’analyse de plusieurs dizaines de mutations en une seule analyse dans un échantillon donné.
À ce jour, l’analyse des biomarqueurs oncologiques à partir d’un fragment de tumeur reste incontournable mais elle pourrait rapidement être optimisée par des analyses réalisées à partir de sang (« biopsie liquide »), plus précisément de cellules tumorales circulantes ou d’ADN tumoral libre circulant. Ces techniques doivent encore être validées en termes de sensibilité, spécificité, faisabilité et impact médico-économique mais l’on peut espérer une application rapide de ces techniques pour le suivi des patients.
Place de l’immunohistochimie dans l’analyse des biomarqueurs
La notion de biomarqueurs oncologiques ne fait pas seulement appel à des techniques moléculaires mais peut également reposer sur l’évaluation de l’expression d’une protéine par les cellules tumorales (par immunohistochimie sur coupes en paraffine). Dans le domaine des CCR, l’immunohistochimie a une place de choix dans l’évaluation de l’expression des protéines MMR. Lorsqu’elle est complète (analyse des quatre protéines MLH1, MSH2, MSH6 et PMS2) et réalisée dans de bonnes conditions, elle est aussi sensible que la biologie moléculaire pour détecter une déficience du système MMR ; elle a de plus l’avantage de cibler la protéine défectueuse et donc d’orienter la recherche de mutation constitutionnelle. Elle représente donc une très bonne alternative à la recherche d’une IMS.
En revanche, l’utilisation de l’immunohistochimie avec l’anticorps VE1, reconnaissant la protéine mutée V600E pour la recherche de mutation BRAF V600E, n’est pas recommandée dans les CCR. En effet, l’analyse des immuno-marquages avec cet anticorps paraît difficile et très variable dans les CCR avec une corrélation très imparfaite avec la mutation BRAF V600E détectée par biologie moléculaire.
Conclusion
Les biomarqueurs actuellement utilisés en pratique clinique concernent d’une part l’IMS et d’autre part la voie EGFR, ses voies connexes et la prédiction de la réponse aux anti-EGFR. Mais, alors que l’approche anti-angiogène, et notamment le bevacizumab (Ac anti-VEGF), fait partie de l’arsenal thérapeutique du CCR métastatique depuis 10 ans on ne dispose toujours pas de facteur prédictif d’efficacité ou de résistance utilisable en pratique clinique.
Actuellement, l’identification des biomarqueurs tumoraux dans le CCR passe par l’étude du niveau d’expression de protéines d’intérêt ou l’étude de la séquence ou de l’amplification des gènes correspondants. De nouvelles approches se développent rapidement et élargissent le champ des biomarqueurs tumoraux ; les petits ARN interférents (ou siRNA pour small interfering RNA) et les signatures d’expression génique ou puces (Oncotype®, Coloprint®) en sont des exemples.
L’oncologie de demain est indissociable du développement des biomarqueurs. La recherche dans le domaine est croissante et ne manquera pas de déboucher sur des améliorations considérables via une meilleure sélection des patients et des traitements. Une meilleure compréhension des mécanismes biologiques tumoraux reste indispensable à l’avancée des biomarqueurs.
Références
- Laurent-Puig P, Agostini J, Maley K. Oncogenèse colorectale. Bulletin du Cancer 2010;97(11):1311-21.
- Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, Tu D, Redston M, Gallinger S. Tumor microsatellite instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003; 349(3):247-57.
- Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, French AJ, Kabat B, Foster NR, Torri V, Ribic C, Grothey A, Moore M, Zaniboni A, Seitz JF, Sinicrope F, Gallinger S. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol 2010;28(20):3219-26.
- Roth AD, Delorenzi M, Tejpar S, Yan P, Klingbiel D, Fiocca R, d’Ario G, Cisar L, Labianca R, Cunningham D, Nordlinger B, Bosman F, Van Cutsem E. Integrated analysis of molecular and clinical prognostic factors in stage II/III colon cancer. J Natl Cancer Inst 2012;104(21):1635-46.
- Gryfe R, Kim H, Hsieh ET, Aronson MD, Holowaty EJ, Bull SB, Redston M, Gallinger S. Tumor microsatellite instability and clinical outcome in Young patients with colorectal cancer. N Engl J Med 2000;342(2):69-77.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23(3):609-18. Review.
- Ogino S, Shima K, Meyerhardt JA, McCleary NJ, Ng K, Hollis D, Saltz LB, Mayer RJ, Schaefer P, Whittom R, Hantel A, Benson AB 3rd, Spiegelman D, Goldberg RM, Bertagnolli MM, Fuchs CS. Predictive and prognostic roles of BRAF mutation in stage III colon cancer: results from intergroup trial CALGB 89803. Clin Cancer Res 2012;18(3): 890-900.
- Gavin PG, Colangelo LH, Fumagalli D, Tanaka N, Remillard MY, Yothers G, Kim C, Taniyama Y, Kim SI, Choi HJ, Blackmon NL, Lipchik C, Petrelli NJ, O’Connell MJ, Wolmark N, Paik S, Pogue-Geile KL. Mutation profiling and microsatellite instability in stage II and III colon cancer: an assessment of their prognostic and oxaliplatin predictive value. Clin Cancer Res 2012;18(23):6531-41.
- Des Guetz G, Schischmanoff O, Nicolas P, Perret GY, Morere JF, Uzzan B. Does microsatellite instability predict the efficacy of adjuvant chemotherapy in colorectal cancer? A systematic review with meta-analysis. Eur J Cancer 2009;45(10):1890-6.
- Milano G, et al. ASCO 2013 abst. 3596
- Sinicrope FA, Foster NR, Thibodeau SN, Marsoni S, Monges G, Labianca R, Kim GP, Yothers G, Allegra C, Moore MJ, Gallinger S, Sargent DJ. DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. J Natl Cancer Inst 2011;103(11): 863-75.
- Kuebler JP, Wieand HS, O’Connell MJ, Smith RE, Colangelo LH, Yothers G, Petrelli NJ, Findlay MP, Seay TE, Atkins JN, Zapas JL, Goodwin JW, Fehrenbacher L, Ramanathan RK, Conley BA, Flynn PJ, Soori G, Colman LK, Levine EA, Lanier KS, Wolmark N. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2007; 25(16):2198-204.
- Patel GS, Karapetis CS. Personalized treatment for advanced colorectal cancer: KRAS and beyond. Cancer Manag Res 2013 Nov 21;5:387-400.
- Mansi L, Viel E, Curtit E, Medioni J, Le Tourneau C. Targeting the RAS signalling pathway in cancer. Bull Cancer 2011;98(9): 1019-28.
- Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006;66(8): 3992-5.
- Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, Bastit L, Killian A, Sesboüé R, Tuech JJ, Queuniet AM, Paillot B, Sabourin JC, Michot F, Michel P, Frebourg T. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer 2007;96(8):1166-9.
- Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, Błasińska-Morawiec M, Šmakal M, Canon JL, Rother M, Williams R, Rong A, Wiezorek J, Sidhu R, Patterson SD. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013;369(11):1023-34.
- Thiel A, Ristimäki A. Toward a Molecular Classification of Colorectal Cancer: The Role of BRAF. Front Oncol 2013 Nov 15;3:281. Review.
- Safaee Ardekani G, Jafarnejad SM, Tan L, Saeedi A, Li G. The prognostic value of BRAF mutation in colorectal cancer and melanoma: a systematic review and meta-analysis. PLoS One 2012;7(10).
- Lièvre A, Rouleau E, Buecher B, Mitry E. Clinical significance of BRAF mutations in colorectal cancer. Bull Cancer 2010;97(12): 1441-52. Review.
- Loupakis F, Cremolini C, Salvatore L, Masi G, Sensi E, Schirripa M, Michelucci A, Pfanner E, Brunetti I, Lupi C, Antoniotti C, Bergamo F, Lonardi S, Zagonel V, Simi P, Fontanini G, Falcone A. FOLFOXIRI plus bevacizumab as first-line treatment in BRAF mutant metastatic colorectal cancer. Eur J Cancer 2014; 50(1):57-63.
- Olschwang S, Bonaïti C, Feingold J, Frébourg T, Grandjouan S, Lasset C, Laurent-Puig P, Lecuru F, Millat B, Sobol H, Thomas G, Eisinger F. Identification and management of HNPCC syndrome (hereditary non polyposis colon cancer), hereditary predisposition to colorectal and endometrial adenocarcinomas. Bull Cancer 2004;91(4): 303-15. Review.
- Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K, Sun R, Nosho K, Meyerhardt JA, Giovannucci E, Fuchs CS, Chan AT, Ogino S. N Engl J med 2012;17: 1596-606.
Les Quatre points forts
- Les biomarqueurs tumoraux utilisés en pratique clinique sont l’instabilité microsatellitaire (IMS) et les mutations de RAS et de BRAF. Leur recherche est réalisée par technique de biologie moléculaire à partir des échantillons tumoraux dans les plateformes hospitalières de génétique moléculaire des cancers mise en place par l’INCa.
-
L’IMS présente dans 15 % des CCR sporadiques est une caractéristique constante des CCR développés dans le cadre d’un syndrome de Lynch. L’IMS est un facteur de bon pronostic des CCR de stade localisé. Les cancers du côlon de stade II avec IMS n’ont pas d’indication à une chimiothérapie adjuvante même s’ils ont des facteurs dits « à haut risque ».
- Une mutation des gènes RASest retrouvée dans 50 à 60 % des CCR. Les mutations RASpeuvent se situer sur les exons 2, 3 ou 4 des gènes KRAS et NRAS. Ce sont des mutations activatrices induisant une résistance aux traitements par Ac anti-EGFR.
- Une mutation du gène BRAFest identifiée dans 5 à 10 % des CCR. Il s’agit presque toujours de la mutation BRAF V600E. Elle est un facteur de mauvais pronostic, surtout au stade métastatique.
FMC HGE : Organisme certifié Qualiopi pour la catégorie ACTIONS DE FORMATION