Aspects hépatiques de la polykystose hépato-rénale
Objectifs pédagogiques
- Connaître les différents types de kystes hépatiques que l’on peut rencontrer dans la polykystose hépatorénale et leur évolution
- Connaître la surveillance de la polykystose hépato-rénale
- Connaître les indications du traitement chirurgical ou radiologique
Introduction
La polykystose hépato-rénale autosomique dominante (PKRAD) est une maladie génétiques fréquente avec une prévalence estimée entre 1/400 et 1/1000 [1]. La mutation génétique PKD1, codant la polycystine-1, est retrouvée dans 80 % des cas, tandis que 20 % des patients sont porteurs de la mutation PKD2, codant la polycystine-2 [2]. Les kystes hépatiques résultent d’une croissance anormale de l’épithélium biliaire (cholangiocytes) ou de la dilatation des glandes péribiliaires, en raison de la persistance de structures biliaires embryonnaires. L’épithélium kystique conserve ainsi les caractéristiques de l’épithélium biliaire, mais avec des activités sécrétoires et prolifératives accrues. Il existe une autre forme de polykystose hépato-rénale beaucoup plus rare, la polykystose rénale autosomique récessive (PKRAR), liée à la mutation PKHD1. Il s’agit d’une forme pédiatrique sévère, survenant pour 1/40 000 naissances, de pronostic rénal défavorable avec un taux de mortalité infantile élevé [3, 4].
Les kystes hépatiques représentent la manifestation extrarénale la plus fréquente, survenant dans plus de 50 % des cas de polykystose hépato-rénale. Ils apparaissent secondairement après l’atteinte rénale, qui fait le pronostic de cette pathologie. Leur prise en charge dépend de la symptomatologie, de l’étendue, de la distribution et de l’anatomie des kystes et peut inclure une aspiration percutanée, une sclérose alcoolique ou une fenestration des kystes ; une hépatectomie partielle voire une greffe de foie sont praticables dans les rares cas où l’hépatomégalie est particulièrement invalidante [5]. Une meilleure connaissance des mécanismes physiopathologiques de la formation des kystes a conduit à proposer les analogues de la somatostatine dans cette indication, mais le bénéfice de ces traitements n’est pas encore clairement établi [6, 7]. Cependant, pour la plupart des patients ayant une polykystose hépato-rénale, le pronostic est bon et aucun traitement n’est nécessaire.
Connaître les différents types de kystes hépatiques que l’on peut rencontrer dans la polykystose hépato-rénale et leur évolution
L’échographie suffit à faire le diagnostic de polykystose hépato-rénale. Dans les formes compliquées, le scanner ou l’imagerie par résonance magnétique (IRM) peuvent être utiles. Les différentes formes radiologiques sont décrites selon la classification de Gigot (Fig. 1) [8].
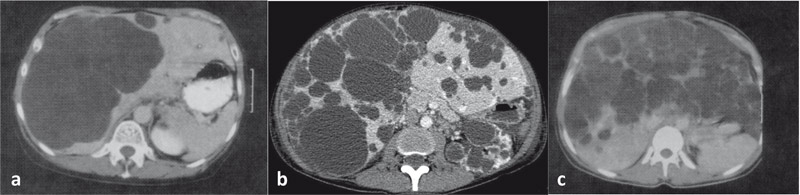 Figure 1. Classification de Gigot (extrait de Gigot JF, et al. Ann Surg 1997;225:286-94)
Figure 1. Classification de Gigot (extrait de Gigot JF, et al. Ann Surg 1997;225:286-94)
Type I (a) : (scanner sans injection) nombre limité (< 10) de larges kystes hépatiques (> 10 cm)
Type II (b) : (scanner temps portal) présence de multiples kystes de taille moyenne avec persistance d'une large zone de parenchyme hépatique sain
Type III (c) : (scanner sans injection) présence de multiples kystes hépatiques de petite et moyenne tailles avec atteinte hépatique diffuse
La présentation clinique traduit la présence de kystes hépatiques de taille (quelques cm à plusieurs dizaines de cm) et de nombres variables. Dans les formes familiales, les critères diagnostiques se définissent par la présence de plus d’un kyste chez les patients de moins de 40 ans, et de plus de trois kystes chez les plus de 40 ans. Pour les patients qui n’ont pas d’antécédents familiaux, le critère retenu est la présence de plus de 20 kystes. Les femmes (79,4 %) sont davantage atteintes que les hommes et ont également un nombre plus important de kystes [9]. Les kystes apparaissent généralement autour de 40 ans (23-84 ans), leur nombre et leur taille augmentent avec l’âge [9]. Des douleurs transitoires de l’hypochondre droit sont fréquentes mais seul un tiers des patients a une symptomatologie chronique ; les symptômes dépendent de la masse et donc de l’effet de compression, et comprennent distension abdominale, satiété précoce, dyspnée et douleurs dorsales liées à l’hépatomégalie [9].
Le fonctionnement hépatique est généralement normal, sans signe d’hypertension portale intrahépatique ni d’insuffisance hépatocellulaire mais l’hépatomégalie peut entraîner une compression cave inférieure et les kystes une compression portale, sus-hépatique ou biliaire. Le bilan biologique s’accompagne alors d’une cholestase anictérique avec une légère augmentation des gGT (inférieure à 3N), voire des phosphatases alcalines dans de rares cas.
Certains kystes, surtout s’ils sont volumineux, peuvent saigner (hémorragie intrakystique ou rupture des kystes), d’autres peuvent se surinfecter. La surinfection de kyste est la complication hépatique la plus redoutée dans la polykystose hépato-rénale. Le diagnostic est idéalement confirmé par la ponction de kyste révélant le germe responsable et une augmentation des polynucléaires neutrophiles dans le liquide. Le plus souvent, le diagnostic probable repose sur les critères suivants : hyperthermie supérieure à 38°C, douleur de l’hypochondre droit, C-réactive protéine supérieure à 5 mg/dl et absence de saignement intrakystique au scanner. L’échographie hépatique peut mettre en évidence des sédiments intrakystiques. Un Pet-scan au FDG (18-fluorodeoxyglucose) peut être proposé en raison de sa supériorité diagnostique prouvée comparativement aux autres techniques d’imagerie [10]. Mais la prise en charge optimale de l’infection de kyste nécessite, outre le drainage du kyste, une double antibiothérapie intraveineuse, si possible adaptée au germe, sinon associant une fluoroquinolone. La durée du traitement est variable selon le terrain et l’efficacité du traitement [11]. L’hémorragie intrakystique est en règle générale limitée et ne menace pas le pronostic vital du patient. Un traitement antalgique symptomatique suffit dans la majorité de cas. Ces complications locales sont plus fréquentes chez les malades ayant une PKRAD avec une insuffisance rénale nécessitant un traitement par dialyse.
Connaître la surveillance de la polykystose hépato-rénale
Dans la polykystose hépato-rénale, le pronostic est clairement lié à l’atteinte rénale. Les complications rénales peuvent apparaître au premier plan sous forme d’une insuffisance rénale chronique, d’une hypertension artérielle, d’une lithiase rénale, d’une pyélo-néphrite aiguë ou d’une hématurie tandis que l’atteinte hépatique succède à l’atteinte rénale, d’une part, et n’engage que très rarement le pronostic vital, d’autre part [12]. Il a été démontré que le pronostic de la maladie rénale était moins bon chez les porteurs de la mutation PKD1 que chez ceux ayant la mutation PKD2 [12]. De plus, il est décrit des kystes plus volumineux et plus nombreux dans les polykystoses hépatiques isolées que dans les poly-kystoses hépato-rénales, par contre les formes sévères sont plus fréquemment observées dans la polykystose hépato-rénale [9]. Les manifestations extra-hépatiques sont également plus fréquentes dans la forme hépato-rénale que dans la forme simple et peuvent inclure des anévrysmes intracrâniens (généralement de petite taille et avec peu de risque de rupture, 17 %) et le prolapsus valvulaire mitral (41 %) [13, 14]. Cependant, il n’est pas recommandé de dépistage systématique de telles anomalies chez les patients porteurs de polykystose hépato-rénale.
Les autres facteurs prédictifs de PKRAD sévère sont la contraception orale ou le traitement œstrogénique substitutif, ainsi que les grossesses multiples [5]. Ces données suggèrent un lien hormonal concourant à éviter tout traitement hormonal substitutif chez les patientes à risque [15].
Il a été décrit une augmentation significative du CA19-9 sérique dans la polykystose hépato-rénale et il n’est pas rare de retrouver des taux sériques équivalents à 3N, en dehors d’une -cholestase sans influence du sexe ni de l’âge [16]. De plus, ce marqueur a été proposé comme marqueur de complication infectieuse des kystes dans la PKRAD, atteignant des taux > 100 000 ng/ml dans le liquide de ponction, mais aucun seuil de significativité n’a été défini dans le sérum [16, 17].
Connaître les indications du traitement chirurgical ou radiologique
Traitement chirurgical
La technique chirurgicale diffère selon qu’il s’agit d’une forme symptomatique de polykystose hépato-rénale à gros kystes (> 5 cm), où l’on privilégie la technique chirurgicale de fenestration sous cœlioscopie, et une forme à petits kystes (< 5 cm) type Gigot III, où une hépatectomie voire la transplantation hépatique (TH) sera proposée, le plus souvent combinée à la transplantation rénale.
1) La fenestration cœlioscopique est un traitement efficace lorsqu’il existe peu de volumineux kystes (Gigot I et II), et qui a l’avantage de pouvoir être répétée en cas de récidive. Cependant, le bilan préopératoire a tendance à surestimer la taille des kystes volumineux qui sont en fait constitués de plusieurs kystes accolés, obligeant à réaliser des fenestrations de proche en proche à travers les kystes superficiels. Dans 92 % des cas, la fenestration laparoscopique apporte une réduction des symptômes mais les kystes récidivent dans 22 % des cas [18]. De plus, les complications sont fréquentes (23 %) : ascite, épanchement pleural, saignement et fuite biliaire. Il est important que la fonction rénale soit préservée sinon il existe un risque d’ascite postopératoire chronique.
2) La fenestration chirurgicale est plus complète que la fenestration cœlioscopique mais elle n’est pas toujours efficace s’il existe de multiples kystes de petite taille. En cas de récidive, la réintervention est difficile. C’est la raison pour laquelle ce traitement a tendance à être abandonné au profit de la résection chirurgicale de la portion hépatique la plus kystique.
3) La résection hépatique est indiquée dans les formes Gigot II sévères avec au minimum un segment hépatique sain ; elle se justifie par le fait que la grande majorité des polykystoses est constituée de multiples kystes de petite taille, impossibles ou difficiles à fenestrer. Comme les kystes sont répartis de façon inhomogène dans le parenchyme avec en permanence une zone de parenchyme épargnée, la sauvegarde de cette zone va permettre une régénération hépatique relativement indemne de kystes avec un résultat durable. Cette intervention est très efficace sur les symptômes (86 %) mais elle reste difficile à réaliser car les plans de scissures anatomiques sont refoulés par les kystes. Cette technique connaît cependant une morbidité (51 %) et une mortalité (3 %) importantes [18]. Les suites de ces interventions sont marquées par une ascite parfois importante associée à un risque d’hémorragie postopératoire et de fuites biliaires dues à l’ouverture de canaux biliaires qui étaient comprimés dans la paroi des kystes. Ces hépatectomies sont mal tolérées chez les malades dénutris et chez ceux ayant une altération de la fonction rénale.
4) La TH peut être isolée, ou associée à une transplantation rénale lorsqu’il existe une polykystose hépato-rénale symptomatique associée à une insuffisance rénale chez un malade dialysé. Ce traitement a en outre l’avantage de faciliter la tolérance immunologique de la transplantation rénale lorsque les deux organes proviennent du même donneur. Les bons résultats de la transplantation rénale tendent à en élargir les indications de la double transplantation aux malades ayant un risque important avec la résection, notamment en cas de dénutrition ou lorsqu’il existe une insuffisance rénale même non dialysée. De façon similaire, une TH isolée est parfois proposée en cas d’hépatectomie large avec un volume résiduel de foie résiduel prévisible trop faible (< 0,5 % ´ poids du corps). La polykystose est considérée comme une exception au score de Meld pour la TH. La survie dans cette indication est excellente (92 %) à 5 ans [19].
Traitement radiologique
La ponction avec injection d’alcool ou de produits sclérosants peut être proposée aux malades ayant un kyste dominant symptomatique, de plus de 5 cm [18]. La ponction a l’avantage d’établir l’imputabilité de la symptomatologie à la présence du kyste. La technique consiste à ponctionner et vider le kyste puis injecter un produit sclérosant (tels que l’alcool ou la minocycline) pour détruire la paroi du kyste et éviter sa reformation. L’efficacité de ce traitement peut se poursuivre après plusieurs mois et une thérapeutique complémentaire ne doit pas être proposée avant 6 mois. Cependant, l’efficacité inconstante et non durable du traitement percutané conduit à lui préférer la fenestration cœlioscopique.
La ponction/drainage de kyste reste la technique de choix en cas de surinfection de kyste. Elle permet d’étayer le diagnostic bactériologique et d’optimiser la prise en charge thérapeutique.
L’embolisation sélective des branches artérielles irriguant les kystes a été proposée mais cette pratique n’a pas encore fait la preuve de son efficacité et reste limitée à des essais cliniques.
L’efficacité des analogues de la somatostatine (Lanreotide et Octreotide) sur la réduction de la taille des kystes hépatiques a été démontrée dans plusieurs études indépendantes mais de petite taille et de durée relativement courte [6, 7].
À ce jour, il n’existe pas de consensus établi sur la prise en charge de la polykystose hépato-rénale mais le schéma thérapeutique décrit sur la figure 2 peut être proposé (Fig. 2) [5].
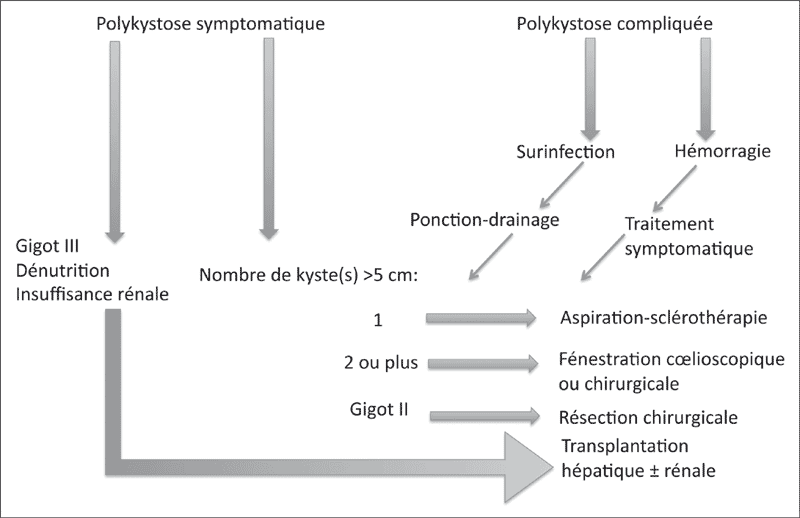 Figure 2. Algorithme de prise en charge radiologique et chirurgicale de la polykystose hépato-rénale
Figure 2. Algorithme de prise en charge radiologique et chirurgicale de la polykystose hépato-rénale
Références
- Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet 2007;369:1287-1301.
- Bergmann C, Weiskirchen R. It’s not all in the cilium, but on the road to it: genetic interaction network in polycystic kidney and liver diseases and how trafficking and quality control matter. J Hepatol 2012;56:1201-3.
- Onori P, Franchitto A, Mancinelli R, Carpino G, Alvaro D, Francis H, et al. Polycystic liver diseases. Dig Liver Dis 2010;42:261-71.
- Büscher R, Büscher AK, Weber S, Mohr J, Hegen B, Vester U, et al. Clinical manifestations of autosomal recessive polycystic kidney disease (ARPKD): kidney-related and non-kidney-related phenotypes. Pediatr Nephrol 2013; [Epub ahead of print].
- Gevers TJG, Drenth JPH. Diagnosis and management of polycystic liver disease. Nat Rev Gastroenterol Hepatol 2013;10:101-8.
- Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3’,5’-cyclic monophosphate. Gastroenterology 2007;132:1104-16.
- Hogan MC, Masyuk TV, Page L, Holmes DR 3rd, Li X, Bergstralh EJ, et al. Somatostatin analog therapy for severe polycystic liver disease: results after 2 years. Nephrol Dial Transplant 2012;27:3532-9.
- Gigot JF, Jadoul P, Que F, Van Beers BE, Etienne J, Horsmans Y, et al. Adult polycystic liver disease: is fenestration the most adequate operation for long-term management? Ann Surg 1997;225:286-94.
- Hoevenaren IA, Wester R, Schrier RW, McFann K, Doctor RB, Drenth JPH, et al. Polycystic liver: clinical characteristics of patients with isolated polycystic liver disease compared with patients with polycystic liver and autosomal dominant polycystic kidney disease. Liver Int 2008;28:264-70.
- Jouret F, Lhommel R, Beguin C, Devuyst O, Pirson Y, Hassoun Z, et al. Positron-emission computed tomography in cyst infection diagnosis in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2011;6:1644-50.
- Sallée M, Rafat C, Zahar J-R, Paulmier B, Grünfeld J-P, Knebelmann B, et al. Cyst infections in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2009;4:1183-9.
- Robinson C, Hiemstra TF, Spencer D, Waller S, Daboo L, Karet Frankl FE, et al. Clinical utility of PKD2 mutation testing in a polycystic kidney disease cohort attending a specialist nephrology out-patient clinic. BMC Nephrol 2012;13:79.
- Ecder T, Schrier RW. Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat Rev Nephrol 2009;5: 221-8.
- Xu HW, Yu SQ, Mei CL, Li MH. Screening for intracranial aneurysm in 355 patients with autosomal-dominant polycystic kidney disease. Stroke 2011;42:204-6.
- Everson GT, Helmke SM, Doctor B. Advances in management of polycystic liver disease. Expert Rev Gastroenterol Hepatol 2008;2: 563-76.
- Kanaan N, Goffin E, Pirson Y, Devuyst O, Hassoun Z. Carbohydrate antigen 19-9 as a diagnostic marker for hepatic cyst infection in autosomal dominant polycystic kidney disease. Am J Kidney Dis 2010;55:916-22.
- Jouret F, Lhommel R, Devuyst O, Annet L, Pirson Y, Hassoun Z, et al. Diagnosis of cyst infection in patients with autosomal dominant polycystic kidney disease: attributes and limitations of the current modalities. Nephrol Dial Transplant 2012;27:3746-51.
- Drenth JPH, Chrispijn M, Nagorney DM, Kamath PS, Torres VE. Medical and surgical treatment options for polycystic liver disease. Hepatology 2010;52:2223-30.
- Van Keimpema L, Nevens F, Adam R, Porte RJ, Fikatas P, Becker T, et al. Excellent survival after liver transplantation for isolated polycystic liver disease: an European Liver Transplant Registry study. Transpl Int 2011;24: 1239-45.
Les Cinq points forts
-
La polykystose hépato-rénale est une maladie génétique fréquente à transmission autosomique dominante, les mutations génétiques PKD1 et PKD2 sont trouvées dans 80 % et 20 % des cas respectivement.
-
Son pronostic est surtout lié à l’atteinte rénale.
-
Elle touche plus les femmes de plus de 40 ans et s’aggrave avec l’âge. Elle est favorisée par la contraception orale, le traitement hormonal substitutif, les multiples grossesses.
-
Le traitement n’est indiqué que pour les kystes symptomatiques. Le traitement chirurgical est préféré au traitement radiologique.
-
L’indication chirurgicale dépend de la taille, de la répartition des kystes, et du volume de foie restant. La fenestration sous cœlioscopie est privilégiée en cas de polykystose à gros kystes (> 5 cm), tandis qu’une résection, voire la transplantation hépatique, seront proposées en cas de forme à petits kystes (< 5 cm).
FMC HGE : Organisme certifié Qualiopi pour la catégorie ACTIONS DE FORMATION