Savoir évoquer une cause génétique devant une cholestase
Connaître les modalités diagnostiques et les indications des tests génétiques
Connaître les modalités thérapeutiques et les récentes innovations pharmacologiques
Savoir gérer la transition des cas pédiatriques en médecine adulte
Les points forts
Les PFIC (Progressive Familial Intrahepatic Cholestasis) sont des maladies chroniques sévères évoluant le plus souvent vers la cirrhose dès l’enfance et nécessitant le recours à la transplantation hépatique.
La PFIC de type 3 peut être d’évolution plus progressive et se révéler à l’âge adulte.
Les BRIC ( Benign Recurrent Intrahepatic Cholestasis) se caractérisent par des épisodes récurrents de cholestase ictérique régressive sans évolution vers la cirrhose.
Le diagnostic des PFIC et des BRIC repose sur un faisceau d’arguments cliniques, biologiques, histologiques et la mise en évidence de variants pathogènes des gènes des transporteurs biliaires.
Le traitement des cholestases génétiques repose principalement sur l’acide ursodésoxycholique et celui du prurit a été récemment amélioré par l’arrivée des inhibiteurs des transporteurs iléaux des acides biliaires (iBAT).
Vidéo
I’m visible only if js is disabled
LIENS D’INTÉRÊT
Consultante pour Ipsen ; Mirum ; Servier ; Amgen ; Gilead. Oratrice pour Ipsen ; Mirum ; Gilead.
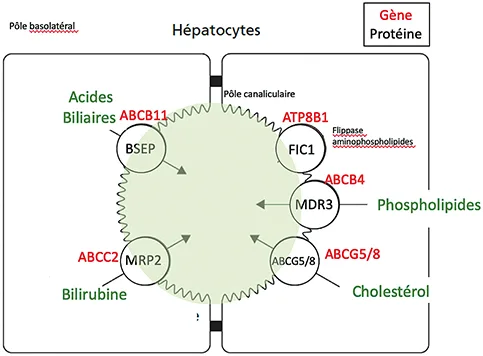
La sécrétion de la bile est une fonction essentielle du foie qui permet la digestion des graisses, l’absorption intestinale des lipides et l’élimination de l’organisme de substances toxiques, endogènes et exogènes. La bile, sécrétée par les hépatocytes, est composée de plusieurs éléments : les acides biliaires, le cholestérol, les phospholipides et la bilirubine. L’excrétion de ces différents éléments par les hépatocytes se fait sous la dépendance de transporteurs situés au pôle canaliculaire des hépatocytes. Les principaux transporteurs sont : le transporteur de la phosphatidylsérine FIC1 (flippase qui assure l’asymétrie de la membrane plasmique lipidique, indispensable au maintien du pôle canaliculaire) codé par le gène ATP8B1 (ATPase Phospholipid Transporting 8B1), le transporteur des acides biliaires BSEP (Bile Salt Export Pump) codé par le gène ABCB11 (ATP-binding cassette subfamilyB member 11) et le transporteur des phospholipides MDR3 (Multidrug Resistance Protein 3) codé par le gène ABCB4 (ATP–binding cassette subfamily B member 4) (figure 1) (1).
Figure 1 : Transporteurs biliaires assurant l’excrétion des différents composants de la bile
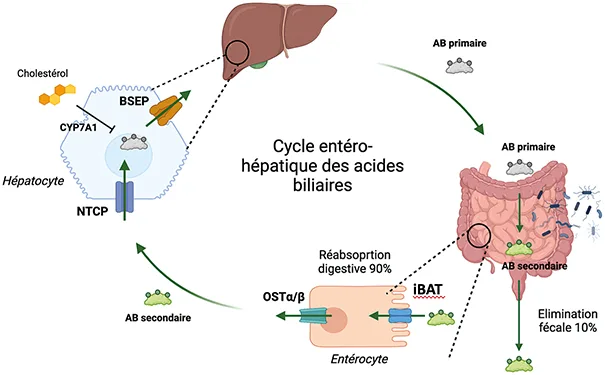
La bile une fois sécrétée au pôle canaliculaire des hépatocytes chemine dans les canalicules biliaires, puis dans les canaux biliaires de petite taille, ensuite dans les gros canaux biliaires avant de rejoindre la lumière intestinale après avoir franchi la papille duodénale. C’est dans la lumière intestinale que les acides biliaires pourront exercer leur fonction de digestion des graisses. La majorité des acides biliaires sont ensuite réabsorbés par les entérocytes sous l’action d’un transporteur spécifique, appelé iBAT (ileal bile acid transporteur) pour être réacheminés vers le foie via la circulation portale. Ce cycle entéro-hépatique permet le recyclage des acides biliaires (figure 2).
Figure 2 : Cycle entéro-hépatique des acides biliaires (AB) : Les acides biliaires (AB) primaires sont synthétisés dans les hépatocytes à partir du cholestérol, par l’action de l’enzyme cholestérol 7 alpha-hydroxylase ou cytochrome P450 7A1 (CYP7A1). Les AB primaires sont ensuite secrétés par les hépatocytes, exportés à leur pôle canaliculaire par le transporteur Bile Salt Export Pump (BSEP). Ils circulent dans les voies biliaires et arrivent dans le duodénum. Dans l’intestin, les AB primaires sont transformés en AB secondaires par les bactéries du microbiote intestinal. Dans l’intestin, 90% des AB sont réabsorbés par le transporteur iléal des acides biliaires (iBAT) situé au pôle apical des entérocytes Les AB sont ensuite exportés dans le sang par le transporteur Organic solute transporter a/b (OSTa/b) situé au pôle basal des entérocytes et regagnent le foie par le système porte. Ils sont recaptés par les hépatocytes par le transporteur Na+-taurocholate cotransporting polypeptide (NTCP). Ce cycle entéro-hépatique permet de recycler les AB. Seulement 10 % des AB synthétisés sont éliminés dans les selles.
La cholestase se définit par une diminution ou un arrêt de la sécrétion biliaire avec pour conséquence une augmentation de la concentration des acides biliaires dans le tissu hépatique. Or ces acides biliaires, de par leurs propriétés détergentes, vont entraîner des lésions des hépatocytes et des cholangiocytes, aboutissant à une inflammation chronique puis à la fibrose hépatique. On distingue les cholestases de cause extra-hépatiques (s’il existe un obstacle sur les gros canaux biliaires, notamment la voie biliaire principale) des cholestases de cause intrahépatique (si la bile n’est pas correctement sécrétée par le tissu hépatique). Parmi les cholestases d’origine intrahépatique se trouvent les cholestases génétiques. Les gènes ATP8B1, ABCB11 et ABCB4 peuvent être le siège de mutations constitutives (germinales ou de novo) entraînant une diminution, voire une perte de fonction du transporteur correspondant (respectivement FIC1, BSEP et MDR3), ce qui va altérer la sécrétion de la bile et entraîner une cholestase intrahépatique. Ces mutations génétiques sont à l’origine d’un groupe de maladies appelées cholestases intrahépatiques familiales. Les cholestases intrahépatiques familiales regroupent des maladies bénignes comme la cholestase récurrente bénigne (Benign Recurrent Intrahepatic Cholestasis, BRIC), le syndrome LPAC (Low Phospholipid-Associated Cholelithiasis), les cholestases d’origine médicamenteuse ou la cholestase gravidique, et des maladies hépatiques plus sévères que sont les cholestases intrahépatiques familiales progressives (Progressive Familial Intrahepatic Cholestasis, PFIC). Cette mise au point sur les cholestases génétiques de l’adulte se focalisera sur les PFIC et les BRIC.
Démarche diagnostique devant une cholestase chez l’adulte
La société européenne d’hépatologie a émis des recommandations sur la démarche diagnostique à adopter devant une cholestase en réalisant les examens complémentaires par étapes de l’examen le moins invasif au plus invasif (figure 3) (2). Devant toute cholestase, il convient en premier lieu de mener un interrogatoire minutieux et un examen clinique afin de collecter des éléments d’orientation, notamment la prise de médicaments. Après avoir éliminé une hépatite virale par la réalisation de sérologies, il convient de faire une échographie hépatique, comme imagerie de première intention à la recherche d’une dilatation des voies biliaires qui serait le témoin d’un obstacle sur les voies biliaires. Si l’échographie ne retrouve pas de dilatation des voies biliaires, il s’agit alors d’une cholestase intrahépatique.
Dans cette situation, l’examen complémentaire à réaliser en première intention sera la recherche des anticorps de la cholangite biliaire primitive (CBP) : anti-mitochondrie, anti-sp100, anti-gp210. Leur positivité associée à une cholestase permet de poser le diagnostic de CBP. Si ces anticorps sont négatifs, il faudra ensuite faire une bili-IRM à la recherche de signes de cholangite sclérosante (sténoses et dilatations des voies biliaires intra et extra-hépatiques) primitive ou secondaire. Si la bili-IRM ne permet pas d’aboutir à un diagnostic, alors la biopsie hépatique sera indiquée notamment pour rechercher une CBP séronégative ou une cholangite sclérosante primitive (CSP) des petits canaux. Si la biopsie hépatique ne permet pas d’aboutir à un diagnostic, il est alors indiqué de faire un génotypage des transporteurs biliaires (ATP8B1, ABCB11 et ABCB4) à la recherche d’une cholestase génétique. La recherche d’une cause génétique est donc recommandée à la toute fin des explorations incluant la biopsie hépatique, quand toutes celles-ci se sont révélées négatives. Néanmoins, dès le départ, certains éléments doivent faire évoquer une origine génétique à la cholestase : un prurit intense, un début des symptômes à un âge jeune, une consanguinité, la présence d’antécédents familiaux de maladie chronique du foie (notamment des cirrhoses de cause indéterminée ou des cancers primitifs du foie), des antécédents personnels et familiaux de pathologies du même spectre (cholestase gravidique, lithiase biliaire intrahépatique) (3). D’autre part, si un diagnostic a été établi pour expliquer la cholestase (par exemple une une CSP des petits canaux) mais que le phénotype initial ou l’évolution sous traitement est inhabituel(le), il est alors recommandé de remettre en cause le diagnostic initial et de s’interroger sur la possibilité d’une cholestase d’origine génétique comme diagnostic différentiel (3). Le génotypage des gènes des transporteurs biliaires se fait désormais par la technique next generation sequencing (NGS) d’un panel de gènes. Il est important de noter que si ce génotypage met en évidence la présence de variants (terme devant désormais être utilisé pour désigner les mutations génétiques), il sera ensuite indispensable de déterminer le caractère pathogène ou non de ces variants, ce qui doit être fait par un généticien expert de ces maladies rares. Il arrive que les variants génétiques retrouvés soient de signification inconnue, ne permettant pas de conclure à un diagnostic de façon formelle. Dans ce cas, il sera nécessaire de ré-analyser les résultats génétiques tous les trois ans, à la lumière des nouvelles connaissances médicales, voire de refaire un nouveau génotypage par une technique plus large comme le whole exome sequencing (WES) qui permet non pas de séquencer un panel de gènes mais l’ensemble de l’exome (3). Dans tous les cas, la discussion entre le clinicien et le généticien est cruciale pour pouvoir conclure ou non à un lien entre le phénotype et le génotype.
PFIC de l’adulte
Les PFIC sont des maladies cholestatiques chroniques rares (prévalence de 1 pour 100 000 habitants), débutant le plus souvent dans les premiers mois de vie et évoluant généralement rapidement vers la cirrhose biliaire secondaire nécessitant le recours à la transplantation hépatique dans l’enfance (4,5). La majorité de ces patients une fois adulte seront donc greffés du foie. Néanmoins, le médecin hépato-gastroentérologue d’adultes doit connaître ces maladies car
il existe des formes atténuées de PFIC, notamment la PFIC 3, dont la progression est plus lente et pour lesquelles le diagnostic peut se faire à l’âge adulte
l’amélioration de la prise en charge thérapeutique de ces patients va permettre d’améliorer leur survie sans transplantation, ce qui rend probable le fait que ces patients atteindront maintenant l’âge adulte avant d’être greffés du foie.
Génétique et physiopathologie
Les PFIC sont des maladies génétiques de transmission autosomique récessive. De façon historique, il a été décrit 3 grands types de PFIC selon le gène affecté : la PFIC de type 1, associée à un défaut du gène ATP8B1, la PFIC de type 2 due à un défaut du gène ABCB11 et la PFIC de type 3 liée à un défaut du gène ABCB4. La PFIC 2 est la plus fréquente chez l’enfant. Chez 60 % des patients atteints de PFIC, le génotypage retrouve un variant d’un de ces 3 gènes, principalement des variants faux-sens ou non-sens à l’état homozygote ou hétérozygote composite, ainsi qu’une large proportion de variants aboutissant à un décalage du cadre de lecture. Ces variants sont associés à une perte de fonction plus ou moins complète du transporteur biliaire correspondant (6). Plus récemment, des variants impliquant d’autres gènes ont été identifiés chez des patients atteints de PFIC : il s’agit de gènes codant pour des protéines de jonctions intercellulaire (comme TJP2) ou des protéines du cytosquelette (comme MYO5B) nécessaires à l’adressage et au maintien des transporteurs biliaires au pôle canaliculaire des hépatocytes ou encore de gènes codant pour des protéines régulant l’expression des transporteurs biliaires et/ou la synthèse des acides biliaires (comme FXR) (5). À ce jour, il existe désormais 12 types différents de PFIC (tableau 1) (5).
Type
Gène
Protéine
PFIC 1
ATP8B1
FIC 1
PFIC 2
ABCB11
BSEP
PFIC 3
ABCB4
MDR3
PFIC 4
TJP2
TJP2
PFIC 5
NR1H4
FXR
PFIC 6
SLC51A
OSTa
PFIC 7
USP53
USP53
PFIC 8
KIF12
KIF12
PFIC 9
ZFYVE19
ZFYVE19
PFIC 10
MYO5B
MYO5B
PFIC 11
SEMA7A
SEMA7A
PFIC 12
VPS33B
VPS33B
Tableau 1 : Les différents types de PFIC
Présentation clinique et démarche diagnostique
Les PFIC se manifestent par une cholestase chronique évoluant vers la fibrose. Le prurit est très souvent présent et invalidant. Dans les PFIC 1 et 2, malgré la cholestase, la GGT reste normale ou subnormale, car la cholestase est d’origine hépatocytaire sans lésion des cholangiocytes (qui sont les principales cellules productrices de GGT) (4,5).
Les PFIC sont pratiquement toujours des maladies à début pédiatrique, d’évolution rapidement progressive vers une cirrhose biliaire secondaire. Seule la PFIC 3 peut se révéler à l’adolescence ou à l’âge adulte. La présentation clinique des PFIC 3 est variable, allant de la cholestase chronique asymptomatique à la cirrhose décompensée (7).
Contrairement aux PFIC 1 et 2, la PFIC de type 3 se présente sous la forme d’une cholestase classique à GGT élevée. En effet, le défaut du transporteur MDR3 entraîne une bile appauvrie en phospholipides, ceci a deux conséquences :
la formation de micelles autour des acides biliaires n’est plus possible et les acides biliaires libres dans la bile vont entraîner des lésions des cellules bordant les canaux biliaires (les cholangiocytes) aboutissant à une cholangite,
cette faible concentration en phospholipides va entraîner une lithogenèse avec l’apparition de calculs de cholestérol.
C’est pourquoi les patients avec une PFIC 3 ont souvent des calculs biliaires intrahépatiques (physiopathologie identique à celle du syndrome LPAC) et ces calculs vont aggraver la cholestase et les lésions de cholangite. En cas de PFIC 3, la biopsie hépatique montre des lésions de cholangite associées à une réaction ductulaire, une ductopénie et une fibrose biliaire d’intensité variable (8). L’analyse immuno-histochimique de MDR3 sur la biopsie hépatique permet parfois de confirmer le déficit d’expression du transporteur sur la membrane des hépatocytes mais il est important de noter que la présence de la protéine ne permet pas d’attester de sa fonction. Toute ductopénie inexpliquée chez l’adulte doit donner lieu à une recherche de variants pathogènes de ABCB4 (2). Ainsi, le diagnostic de la PFIC 3 repose sur un faisceau d’arguments en faveur, incluant les résultats de l’histologie hépatique, l’élimination des diagnostics différentiels et le génotypage du gène ABCB4.
Pronostic
L’évolution de la PFIC 3 est très variable et dépend du génotype des patients. Une étude française incluant 38 patients atteints de PFIC 3 a montré que la présence d’au moins un variant faux-sens était associée à une meilleure survie sans transplantation et à une meilleure réponse au traitement (9). Les patients atteints de PFIC 3 sont exposés au risque de cirrhose et à ses complications (insuffisance hépatique, hypertension portale, carcinome hépatocellulaire), ainsi qu’au risque de cholangiocarcinome.
Traitement
Pour les patients atteints de PFIC, le traitement de première intention est l’acide ursodésoxycholique (AUDC) qui permet d’améliorer la cholestase mais son efficacité sur la survie sans transplantation n’a pas été démontrée (10).
Le prurit est le plus souvent un symptôme invalidant pour ces patients et doit être pris en charge de façon adaptée. Les traitements du prurit comprennent : la rifampicine, la cholestyramine et le bézafibrate (3). Récemment, de nouvelles molécules ont été démontrées comme efficaces pour diminuer le prurit des patients atteints de PFIC. Il s’agit des inhibiteurs des iBAT, qui empêchent la réabsorption iléale des acides biliaires et donc interrompent le cycle entéro-hépatique des acides biliaires (figure 2). Ces médicaments agissent de la même façon que la dérivation biliaire chirurgicale, traitement qui a longtemps été utilisé pour traiter le prurit des patients avec PFIC. Actuellement, deux inhibiteurs d’iBAT ont obtenu une autorisation de mise sur le marché pour traiter les patients atteints de PFIC : l’odévixibat et le maralixibat. Des essais randomisés contrôlés ont montré que ces deux molécules amélioraient le prurit ainsi que la concentration sérique des acides biliaires (qui est un marqueur pronostique des PFIC chez les enfants) (11,12,13). Cependant, l’efficacité des inhibiteurs d’iBAT sur le prurit est variable d’un patient à l’autre et dépend du génotype. L’effet secondaire principal de ces nouveaux traitements est la diarrhée, qui affecte 30 à 60% des patients et s’explique par une augmentation de la concentration des acides biliaires dans le colon, entraînant un effet laxatif. Cette diarrhée peut être améliorée par une réduction de la posologie des inhibiteurs des iBAT. L’effet des inhibiteurs d’iBAT sur l’évolution de la maladie et la fibrose hépatique n’a pas encore été évalué.
Aujourd’hui, la transplantation hépatique reste le seul traitement curatif des PFIC. Comme dans les autres causes d’hépatopathies chroniques, elle est indiquée en cas d’ictère persistant, de cirrhose décompensée ou de carcinome hépatocellulaire, mais dans un contexte de PFIC, elle est également indiquée en cas de prurit réfractaire au traitement médical.
BRIC de l’adulte
Les cholestases intrahépatiques récurrentes bénignes (benign recurrent intrahepatic cholestasis ou BRIC) font partie du même groupe de maladies génétiques rares que les PFIC. Contrairement aux PFIC, les BRIC sont caractérisées par des épisodes récurrents de cholestase ictérique avec régression complète des symptômes entre les épisodes (14). La prévalence de BRIC est inconnue.
Génétique et physiopathologie
II existe deux types de BRIC : le type 1, associé aux variants d’ATP8B1, et le type 2 associé aux variants de ABCB11. La BRIC se transmet sur un mode autosomique récessif avec une pénétrance incomplète. Le génotypage permet de retrouver un variant pathogène d’ATP8B1 et d’ABCB11 dans, respectivement, 50 % et 25 % des cas (15). Des études plus récentes ont montré que la présence d’un variant d’ATP8B1 ou de ABCB11 à l’état hétérozygote pouvait entraîner un phénotype de BRIC (3). Chez un quart des patients avec un phénotype de BRIC, aucun variant des gènes ATP8B1 ni ABCB11 n’est retrouvé, suggérant qu’il existe d’autres gènes impliqués dans la physiopathologie de ces maladies, comme ce qui a été démontré pour les PFIC. Les variants présents chez les patients atteints de BRIC sont souvent moins sévères que les variants retrouvés chez les patients atteints PFIC 1 et 2 (16), ceci explique que l’évolution des BRIC soit « bénigne », sans évolution vers la cirrhose.
Présentation clinique et démarche diagnostique
L’âge de survenue du premier épisode d’ictère est variable, souvent entre la 2e et la 3e décennie (14). Les épisodes de BRIC peuvent être déclenchés par des facteurs exogènes entraînant une diminution de la fonction des transporteurs biliaires comme une infection bactérienne ou virale, la prise de médicaments (œstro-progestatifs, tétracycline et amoxicilline-acide clavulanique), ou encore une grossesse ou une intervention chirurgicale (17). Au cours des épisodes de BRIC, le prurit qui précède généralement l’ictère est intense et invalidant, retentissant de façon significative sur la qualité de vie du patient. Il existe fréquemment des signes associés, notamment une diarrhée par stéatorrhée secondaire à la cholestase et un amaigrissement important. Les examens biologiques montrent une cholestase ictérique avec élévation de la bilirubine conjuguée et des phosphatases alcalines avec une GGT normale ou subnormale, comme dans les PFIC 1 et 2 (cf. supra). L’élévation des transaminases est variable. La durée moyenne des épisodes de cholestase est d’environ 3 mois, mais elle peut varier de 2 à 24 mois (14). L’intervalle libre entre les crises est aussi très variable, avec une moyenne de 2 ans entre chaque crise. Du fait de l’expression extra-hépatique de la protéine FIC1, les patients atteints de BRIC 1 peuvent présenter des signes extra-hépatiques pendant la poussée de BRIC comme une surdité, une diarrhée, une pneumopathie ou une pancréatite.
La confirmation diagnostique de BRIC nécessite d’exclure les autres causes de cholestase, selon les recommandations européennes (figure 3). En cas de BRIC, la biopsie hépatique montre une cholestase hépatocytaire le plus souvent centrolobulaire isolée, sans fibrose sans cholangite (canaux biliaires normaux) et avec une inflammation absente ou minime et limitée à l’espace porte (18).
La confirmation diagnostique repose sur le génotypage des transporteurs biliaires mais un quart des patients ayant un phénotype de BRIC n’ont pas de variant pathogène identifié d’ATP8B1 et d’ABCB11. Avant l’avènement des tests génétiques, le diagnostic de BRIC devait reposer sur au moins 2 épisodes typiques de cholestase avec régression complète après les épisodes. Aujourd’hui, le diagnostic peut être posé dès le premier épisode si le tableau est typique et si un variant pathogènes des gènes ATP8B1 ou ABCB11 est identifié.
Traitement
Le traitement des épisodes de BRIC repose sur le traitement du prurit. Le traitement médicamenteux comprend la rifampicine (150 à 600 mg/j) (19), la cholestyramine (20) et le bézafibrate (3). Le traitement par AUDC n’a pas fait preuve de son efficacité, ni pour réduire le prurit, ni pour réduire la durée de la poussée de BRIC, ni pour prévenir le risque de récidive.
Le traitement médicamenteux du prurit est souvent insuffisant pour améliorer les patients en poussée de BRIC. Dans ce cas, les traitements invasifs sont indiqués tels que la pose d’un drain nasobiliaire par voie endoscopique (qui permet un drainage externe de la bile et interrompt le cycle entéro-hépatique des acides biliaires). Le drainage nasobiliaire est souvent très efficace pour diminuer rapidement le prurit et réduire la durée de la poussée (21) mais il expose à un risque de 30 % de pancréatite iatrogène lors de sa pose (22).
Les inhibiteurs d’iBAT, en interrompant eux aussi le cycle entéro-hépatique, pourraient avoir un intérêt pour traiter les poussées de BRIC, comme récemment rapporté dans une série de cas cliniques (23). Les autres traitements invasifs du prurit sont les échanges plasmatiques ou la dialyse sur colonne d’albumine (système MARS®) (3).
Pronostic
Contrairement aux PFIC, les BRIC sont de bon pronostic sans évolution vers la fibrose ni la cirrhose. Entre les épisodes, les patients sont asymptomatiques et leurs tests hépatiques sont normaux ou peu perturbés (14). Chez l’enfant, de rares cas de BRIC évoluant vers un phénotype de PFIC ont été rapportés (24). Seules des poussées fréquentes ou prolongées, avec un prurit invalidant non contrôlables, ayant un retentissement majeur sur la qualité de vie peuvent faire discuter l’indication de transplantation hépatique chez ces patients. De tels cas sont exceptionnels dans cette pathologie déjà rare.
Conclusion
Les BRIC et PFIC sont des maladies cholestatiques rares liées à un déficit de fonction des transporteurs biliaires. Les PFIC évoluent vers la cirrhose, le plus souvent à l’âge pédiatrique. La PFIC 3 est la seule PFIC pouvant se révéler à l’âge adulte. Les BRIC se caractérisent par des épisodes de cholestase ictérique à GGT normale avec un prurit invalidant. Leur évolution est bénigne, sans progression vers la fibrose hépatique. En l’absence de traitement médical curatif, le traitement des PFIC et des BRIC repose largement sur l’acide ursodésoxycholique et sur le traitement du prurit qui a été amélioré par l’arrivée de nouvelles molécules : les inhibiteurs des iBAT.
Le diagnostic de ces maladies rares doit s’appuyer sur l’expertise des centres de référence et centres de compétence des réseaux nationaux Atrésie des Voies biliaires et Cholestases Génétiques (AVB-CG) et Maladies Inflammatoires des Voies Biliaires et Hépatites auto-immunes (MIVB-H) de la filière des maladies rares du foie de l’enfant et de l’adulte FILFOIE (https://www.filfoie.com).
Remerciements
À Pierre-Antoine Soret pour ses conseils et la réalisation des figures 2 et 3.
À Christophe Corpechot et Olivier Chazouillères pour la transmission de leur expertise.
Références
Boyer JL et al. Bile formation and secretion. Compr Physiol. 2013 July ; 3(3): 1035–107
EASL Clinical Practice Guidelines on sclerosing cholangitis. J Hepatol 2022;77(3):761-806.
EASL Clinical Practice Guidelines on genetic cholestatic liver diseases. J Hepatol. 2024 Aug;81(2):303-325.
Baker A et al. Systematic review of progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol 2019;43(1):20-36.
Vitale et al. Genotypes and different clinical variants between children and adults in progressive familial intrahepatic cholestasis: a state-of-the-art review. Orphanet J Rare Dis 2025. 20(1):80.
Delaunay J et al. A functional classification of ABCB4 variations causing progressive familial intrahepatic cholestasis type 3. Hepatology 2016;63(5):1620-31.
Stättermayer AF et al. Variants in ABCB4 (MDR3) across the spectrum of cholestatic liver diseases in adults. J Hepatol 2020;73(3):651-63.
Ziol M et al. ABCB4 heterozygous gene mutations associated with fibrosing cholestatic liver disease in adults. Gastroenterology 2008;135(1):131-41.
Gonzales E et al. Outcomes of 38 patients with PFIC3: Impact of genotype and of response to ursodeoxycholic acid therapy. JHEP Rep 2023;5(10):100844.
Jacquemin E et al. Ursodeoxycholic acid therapy in pediatric patients with progressive familial intrahepatic cholestasis. Hepatology 1997;25(3):519-23.
Van Wessel DBE et al. Genotype correlates with the natural history of severe bile salt export pump deficiency. J Hepatol 2020;73(1):84-93.
Thompson RJ et al. Odevixibat treatment in progressive familial intrahepatic cholestasis: a randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol 2022; 7(9):830-842
Miethke AG et al. Maralixibat in progressive familial intrahepatic cholestasis (MARCH-PFIC): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol 2024; 9(7):620-631.
Luketic VA et al. Benign recurrent intrahepatic cholestasis. Clin Liver Dis 2004;8(1):133-49.
Van Mil SWC et al. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology 2004;127(2):379-84.
Folmer DE et al. Differential effects of progressive familial intrahepatic cholestasis type 1 and benign recurrent intrahepatic cholestasis type 1 mutations on canalicular localization of ATP8B1. Hepatology 2009;50(5):1597-605.
Halawi A et al. Triggers of benign recurrent intrahepatic cholestasis and its pathophysiology: a review of literature. Acta Gastroenterol Belg 2021;84(3):477-86.
Brenard R et al. Benign recurrent intrahepatic cholestasis. A report of 26 cases. J Clin Gastroenterol 1989;11(5):546-51.
Helgadottir H et al. Improvement of cholestatic episodes in patients with benign recurrent intrahepatic cholestasis (BRIC) treated with rifampicin. A long-term follow-up. Scand. J Gastroenterol 2023;58(5):512-20.
Uegaki S et al. Successful treatment with colestimide for a bout of cholestasis in a Japanese patient with benign recurrent intrahepatic cholestasis caused by ATP8B1 mutation.Intern Med 2008;47(7):599-602.
Stapelbroek JM et al. Nasobiliary drainage induces long-lasting remission in benign recurrent intrahepatic cholestasis. Hepatology 2006;43(1):51-3.
Hegade VS et al. The safety and efficacy of nasobiliary drainage in the treatment of refractory cholestatic pruritus:a multicentre European study. Aliment Pharmacol Ther 2016;43(2):294-302.
Di Giorgio et al. Odevixibat for Episodic Intrahepatic Cholestasis due to Biallelic Mutations in ATP8B1: A Case Series. Liver Int 2025;45(8):e70216.
Van Ooteghem NAM et al. Benign recurrent intrahepatic cholestasis progressing to progressive familial intrahepatic cholestasis: low GGT cholestasis is a clinical continuum. J Hepatol 2002;36(3):439-43.
Toute reproduction ou réécriture, totale ou partielle, sans l’accord préalable écrit de la FMC HGE est interdite.
FMC HGE : Organisme certifié Qualiopi pour la catégorie ACTIONS DE FORMATION.
Nous vous invitons à tester vos connaissances sur l’ensemble des QCU tirés des exposés des différents POST’U. Les textes, diaporamas ainsi que les réponses aux QCM seront mis en ligne à l’issue des prochaines journées JFHOD.