Introduction
La douleur abdominale est l’un des motifs de consultation les plus fréquents en gastroentérologie (1). Elle peut poser des problèmes diagnostiques, car beaucoup d’affections digestives sont susceptibles de se révéler par une douleur abdominale et certaines douleurs à projection abdominale sont des douleurs d’origine non digestive. L’amélioration de la douleur, demande première du malade, demeure souvent un problème thérapeutique.
L’analyse d’une douleur à projection abdominale doit amener à s’interroger sur deux points : s’agit- il d’une douleur réellement d’origine digestive ? La douleur viscérale a-t-elle une autre composante, pariétale ou musculaire, nécessitant une prise en charge spécifique ?
Certaines douleurs de topographie abdominale sont des douleurs d’origine non digestive (Table I). En règle générale, ces douleurs sont plutôt aiguës. Certaines pathologies non digestives peuvent néanmoins occasionner des douleurs plus chroniques [2].
![Table I. Affections non digestives pouvant donner des douleurs à projection abdominale, le plus souvent aiguës. Référence [2]](data:image/svg+xml;base64,PHN2ZyB4bWxucz0iaHR0cDovL3d3dy53My5vcmcvMjAwMC9zdmciIHdpZHRoPSI1NjAiIGhlaWdodD0iMjI1IiB2aWV3Qm94PSIwIDAgNTYwIDIyNSI+PHJlY3Qgd2lkdGg9IjEwMCUiIGhlaWdodD0iMTAwJSIgc3R5bGU9ImZpbGw6I2NmZDRkYjtmaWxsLW9wYWNpdHk6IDAuMTsiLz48L3N2Zz4=)
Table I. Affections non digestives pouvant donner des douleurs à projection abdominale, le plus souvent aiguës. Référence [2]
Rappels d’anatomie de l’innervation de la paroi abdominale
La carte des dermatomes de la paroi abdominale antéro-latérale est presque identique à celle de la distribution des nerfs périphériques. L’exception se produit au niveau de L1 où la branche antérieure de L1 bifurque en nerfs ilio-hypogastrique et ilio-inguinal. Chaque dermatome commence postérieurement, au-dessus du foramen intervertébral par lequel le nerf spinal émerge de la colonne vertébrale et suit le trajet des côtes, autour du tronc. Le dermatome T10 contient l’ombilic, alors que le dermatome L1 inclut le pli inguinal.
La peau et les muscles de la paroi antéro-latérale de l’abdomen sont généralement innervés par les nerfs suivants (fig. 1) :

Figure 1. Innervation de la paroi abdominale : vue antérieure
- Les nerfs thoraco-abdominaux – les parties distales, abdominales, des branches antérieures des six derniers nerfs spinaux (T7 à T11) ; ce sont les nerfs intercostaux inférieurs, distalement au rebord costal.
- Les branches cutanées latérales (thoraciques) : des nerfs spinaux T7-T9 ou T10.
- Le nerf subcostal : la grosse branche ventrale du nerf spinal T12.
- Les nerfs ilio-hypogastrique et ilio-inguinal sont les branches terminales de la branche ventrale du nerf spinal L1.
Les nerfs thoraco-abdominaux passent inféro-antérieurement, depuis les espaces intercostaux pour cheminer dans le plan neurovasculaire entre les muscles oblique interne et transverse, et se distribuer à la peau et aux muscles de l’abdomen. Les branches cutanées latérales émergent de la musculature de la paroi antéro-latérale pour pénétrer dans le tissu sous-cutané, le long de la ligne axillaire antérieure (sous la forme de branches antérieures et postérieures) alors que les branches cutanées de la paroi abdominale antérieure percent la gaine du droit pour entrer dans le tissu sous-cutané à courte distance du plan médian. Parmi les branches cutanées antérieures des nerfs thoraco-abdominaux :
- Celles de T7 -T9 se distribuent à la peau située supérieurement à l’ombilic.
- Celles de T10 innervent la peau autour de l’ombilic.
- Celles de T11, avec les branches cutanées du nerf subcostal (T12), ilio-hypogastrique et ilio-inguinal (1) se distribuent à la peau inférieure à l’ombilic.
Au cours de leur trajet à travers la paroi abdominale, les nerfs thoraco-abdominaux, subcostal et ilio- hypogastrique s’anastomosent entre eux.
Les muscles de la paroi antéro-latérale de l’abdomen reçoivent une innervation multisegmentaire par les branches antérieures des nerfs spinaux thoraciques inférieurs (T7-T12) et L1. Les branches se rendent séparément aux muscles (c’est-à-dire sans s’unir en nerfs périphériques multisegmentaires comme cela se produit dans les membres), sous la forme de cinq nerfs thoraco-abdominaux (T7-T11), un nerf subcostal (T12), les nerfs ilio-hypogastrique et ilio-inguinal (L1) qui cheminent dans le plan entre les deuxième et troisième couches. Les branches cutanées latérales se distribuent à la peau latérale à la ligne médio-costale ; les branches cutanées antérieures se rendent à la peau médiale à la ligne médio-costale. Excepté pour L1, les cartes des dermatomes abdominaux et des nerfs périphériques sont identiques. Les dermatomes de repérage sont le dermatome T10, qui inclut l’ombilic et le dermatome L1 qui contient le pli inguinal.
Douleurs d’origine radiculaire
L’irritation des racines thoraciques
Les racines nerveuses peuvent être irritées à leur sortie de la colonne vertébrale. S’il s’agit des dernières vertèbres dorsales ou des lombaires, le trajet douloureux est celui d’une ceinture abdominale, complète ou partielle. Les rappels anatomiques, que nous avons effectués précédemment, nous permettent assez facilement d’identifier la ou les racines nerveuses impliquées dans le déclenchement de la radiculalgie. Les arguments pour une origine vertébrale sont le réveil par les inflexions de la colonne, la sensibilité anormale des tissus de la paroi (pincez-les entre vos doigts, sans presser sur les organes dessous), la correspondance de la douleur abdominale avec un point vertébral du même côté, la morphologie favorisante (femme souvent assez forte et cambrée, ceinture abdominale relâchée). Il n’y a pas de troubles digestifs (la douleur n’est pas rythmée par les repas, il n’y a ni diarrhée ni constipation), pas de pertes génitales ni de brûlures mictionnelles, pas de lien entre la douleur et le cycle menstruel.
Il y a autant d’étiologies de radiculalgies thoraco-abdominales que de causes de lombo-sciatalgies. Les examens d’imagerie et en particulier les radiographies du rachis dorso-lombaire de face, de profil et de ¾ ainsi que du bassin de face en charge, la tomodensitométrie lombaire et l’IRM lombaire permettront ainsi d’identifier les discopathies dégénératives avec ou sans hernie discale, l’arthrose inter-apophysaire postérieure, les kystes synoviaux inter-apophysaires, les séquelles de maladie de Scheuerman dorsale, certains spondylolisthesis, certains tassements vertébraux ostéoporotiques, les pathologies tumorales vertébrales, les atteintes inflammatoire des articulations inter-apophysaires postérieures dans un contexte de rhumatisme inflammatoire axial. Il n’est bien entendu pas possible ici de détailler chacune de ces pathologies, mais un avis rhumatologique s’impose dès qu’une étiologie radiculaire est envisagée, même si les anomalies fréquentes de l’imagerie ne sont pas toujours explicatives des douleurs radiculaires.
Les dérangements intervertébraux mineurs
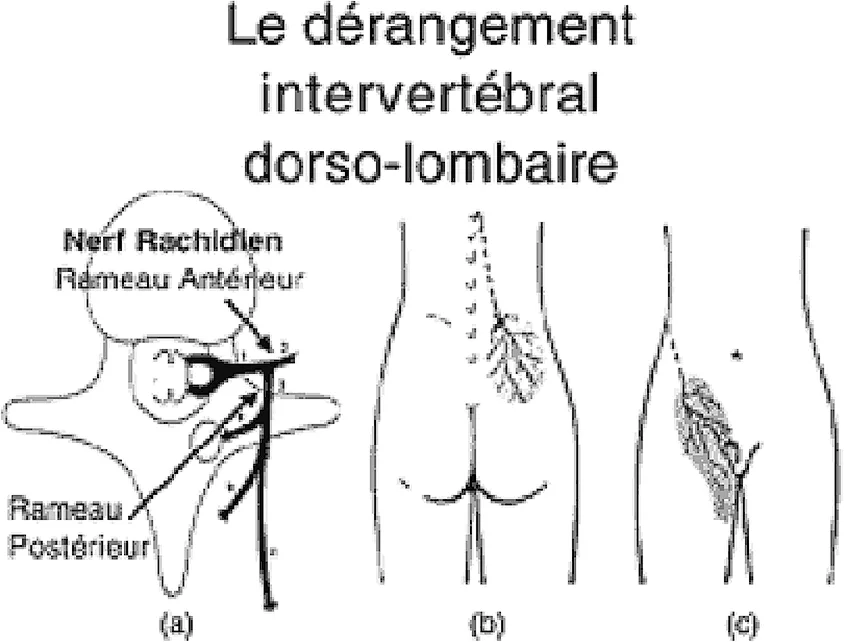
Les dérangements intervertébraux mineurs (DIM), en particulier de la charnière dorso-lombaire, irritent la branche antérieure et/ou postérieure du nerf vertébral et sont fréquemment responsables de douleurs projetées trompeuses. Ils peuvent simuler une pathologie digestive quand la branche antérieure du nerf rachidien est concernée par ce trouble de la statique vertébrale. La symptomatologie peut être favorisée par une recrudescence de l’activité professionnelle ou physique, une affection intercurrente, sources de décompensation d’un dérangement intervertébral mineur jusque-là muet. L’atteinte de la branche antérieure déclenche une gêne ou une véritable douleur, le plus souvent unilatérale, de topographie iliaque (fig. 3). Au niveau de la fosse iliaque, la technique d’examen dite du “pincé-roulé” retrouve une zone cellulalgique. Le diagnostic est renforcé par la découverte d’une atteinte également du rameau postérieur du nerf rachidien à l’origine d’une cellulalgie de la fesse et d’une douleur à la palpation appuyée de la crête iliaque (“point de crête”). Un geste local (infiltration ou manipulation vertébrale) permet souvent une régression de la douleur [3].

Figure 3. Le dérangement inter-vertébral dorso-lombaire. (a) Branches du nerf rachidien qui est impliqué dans la survenue des phénomènes douloureux. (b) Projection douloureuse en cas d’atteinte du rameau postérieur du nerf. (c) Projection douloureuse en cas d’atteinte du rameau antérieur du nerf [2]
Douleurs d’origine pariétale et musculaire
L’existence d’une composante pariétale pourrait s’observer dans 10 à 15 % des douleurs abdominales. Elle est souvent méconnue, cette méconnaissance conduisant à de nombreux examens complémentaires inutiles. La douleur est due notamment à une atteinte des nerfs cutanés abdominaux, en particulier celle de leur branche antérieure qui, dans son trajet au niveau du bord externe du grand droit, traverse une gaine fibreuse où le rameau nerveux peut être traumatisé. Le conflit peut être favorisé par l’augmentation de la pression intra-abdominale (fig. 4). Une composante pariétale est suspectée devant une zone localisée particulièrement douloureuse, notamment lors de la toux et des mouvements. L’existence d’un signe de Carnett est un élément clé pour le diagnostic : la flexion de la tête sur le tronc majore la douleur lors de la pression avec un doigt de la zone douloureuse, alors que cette manœuvre s’associe plutôt à une diminution de la douleur en cas de douleur viscérale. L’injection d’un mélange anesthésique-corticoïde améliore la douleur pariétale dans 60 à 90 % des cas. Le nombre d’injections nécessaire pour obtenir un effet symptomatique peut varier de 1 à 6.
Les hernies de Spiegel se développent à travers la ligne semi-lunaire, au bord externe du muscle grand droit, souvent au-dessous de ligament arqué qui se situe en général à mi-distance entre l’ombilic et le pubis. Le diagnostic est facile, lorsqu’existe à ce niveau une tuméfaction impulsive à la toux. L’examen peut être plus difficile en cas de surcharge pondérale. Surtout la hernie est souvent intra-pariétale. L’échographie pariétale est alors utile au diagnostic.
La douleur abdominale peut avoir également une composante musculaire. La contracture du muscle psoas donne une douleur iliaque uni ou bilatérale, souvent accentuée à la poussée. La palpation de la fosse iliaque identifie un muscle psoas contracté et douloureux. Point important pour le diagnostic : la douleur se majore lors de la flexion de la cuisse sur l’abdomen. L’étude prospective de 74 malades consécutifs ayant un syndrome de l’intestin irritable défini par les critères de Rome II nous a montré que ce spasme n’était pas exceptionnel, puisqu’il a été trouvé chez près d’un malade sur deux. Il pourrait être favorisé par certains mouvements de gymnastique comme la pratique d’abdominaux où le psoas est sollicité pendant près des deux tiers du mouvement. [2]
![Figure 4. Mécanisme d’une douleur pariétale par conflit du rameau antérieur d’un nerf cutané abdominal au niveau de sa gaine fibreuse au bord du muscle grand droit de l’abdomen [2]](data:image/svg+xml;base64,PHN2ZyB4bWxucz0iaHR0cDovL3d3dy53My5vcmcvMjAwMC9zdmciIHdpZHRoPSI0NTAiIGhlaWdodD0iMzU1IiB2aWV3Qm94PSIwIDAgNDUwIDM1NSI+PHJlY3Qgd2lkdGg9IjEwMCUiIGhlaWdodD0iMTAwJSIgc3R5bGU9ImZpbGw6I2NmZDRkYjtmaWxsLW9wYWNpdHk6IDAuMTsiLz48L3N2Zz4=)
Figure 4. Mécanisme d’une douleur pariétale par conflit du rameau antérieur d’un nerf cutané abdominal au niveau de sa gaine fibreuse au bord du muscle grand droit de l’abdomen [2]
Les douleurs en regard du grill costal inférieur : syndrome de Cyriax, Syndrome de Tietze et autres xyphodynies
Le syndrome de Cyriax est une affection mal connue des cliniciens, décrite pour la première fois en 1919 par E.F. Cyriax, chirurgien orthopédiste anglais. Il s’agit d’une douleur liée à une subluxation du cartilage des extrémités antérieures d’une des dernières côtes (8e, 9e ou 10e) qui emprisonne le nerf intercostal sus- jacent (Fig. 5). La compression de ce dernier, lors de certains mouvements ou de certaines postures, déclenche la douleur. D’autres appellations ont suivi, côte glissante (slipping rib), côte claquante (clicking rib), syndrome de l’extrémité costale (rib tip syndrome) ou encore syndrome de la côte douloureuse (painful rib syndrome). L’incidence de ce syndrome lors d’une consultation pour douleurs abdominales chroniques est de 1,5 % dans la série de Sanou et al. [4] et 1 % dans la série de Monnin et al. [5]. Sur le plan épidémiologique, on trouve un sex-ratio de 2 à 3 en faveur des femmes et une moyenne d’âge variant de 47,5 à 50,6 ans selon les auteurs. Sur 100 observations, un traumatisme direct est trouvé dans 71 cas et indirect dans 21 [6]. La symptomatologie est une douleur en ceinture, basi-thoracique et de caractère mécanique, déclenchée par les mouvements du tronc et les efforts, notamment de toux. Il s’agit d’une douleur initialement aiguë, qui peut devenir chronique en cas de retard diagnostique. Elle évolue souvent depuis plusieurs mois, entre 8 et 41 mois selon les séries, avant que le diagnostic ne soit fait.
L’atteinte est le plus souvent unilatérale à droite (67 % à 75 % des cas) et localisée à la dixième côte par traumatisme direct (coup de poing, chute, accident de la voie publique), indirect (traction sur les muscles intercostaux et abdominaux, notamment lors de certains sports comme la gymnastique, le golf, ou le tennis) [7], ou microtraumatismes répétés. Un claquement est parfois ressenti par le patient, lorsque le cartilage luxé reprend sa place. L’examen clinique utilise la manœuvre du crochetage, permettant avec le doigt positionné en dessous du rebord costal de déclencher une douleur en tirant vers le haut, ce qui a pour conséquence de pincer le nerf ; cet examen est quasi pathognomonique du syndrome. E.M. Scott et al. [8] proposent même une triade diagnostique : une douleur basi-thoracique ou épigastrique, un point douloureux exquis du rebord costal et une reproduction de la douleur par la manœuvre du crochetage. Le syndrome de Cyriax pouvant évoquer de nombreuses affections thoraciques, abdominales et/ou viscérales, est fréquemment à l’origine d’une longue errance diagnostique. Dans un tiers des cas les douleurs peuvent être étiquetées d’origine psychogène et conduire à une évaluation psychologique [9]. Parmi les diagnostics différentiels pariétaux, on retrouve le syndrome de Tietze (inflammation douloureuse des articulations chondrocostales supérieures et sternoclaviculaires), la xyphoidalgie (douleur sternale déclenchée par les mouvements, reproduite par la pression sur l’appendice xiphoïde) et la compression des nerfs cutanés abdominaux par leur trajet dans la gaine fibreuse du muscle grand droit (l’existence d’un signe de Carnett est un élément clé du diagnostic d’atteinte pariétale). Les examens paracliniques sont inutiles la plupart du temps, à l’exception de la radiographie du gril costal, voire d’une tomodensitométrie à la recherche de fracture ou de microfissures de la côte concernée. Meuvly et al. [10] recommandent l’utilisation de l’échographie visualisant la mobilité anormale du cartilage avec subluxation lors de la contraction des muscles abdominaux.
Cependant, le faible nombre de cas relatés dans la littérature ainsi que la nécessité d’une formation spécifique restreint l’indication de cet examen. L’attitude thérapeutique est loin d’être codifiée. La plupart des auteurs conseillent de traiter en fonction de l’intensité des douleurs. Dans les douleurs modérées, il est conseillé le port d’un demi-bandage élastique pendant 1 à 2 semaines, associé à des antalgiques per os, ainsi qu’un soutien psychologique [11]. Dans les douleurs vives, l’infiltration locale de corticoïdes plus ou moins associée à de la lidocaïne 2 % fait céder immédiatement la douleur, la guérison pouvant nécessiter plusieurs infiltrations (de 1 à 6). L’amélioration de la douleur sur le long terme varie entre 60 et 90 % des cas. L’échec du traitement médical peut conduire au traitement chirurgical qui consiste en la résection des 5 à 8 derniers centimètres du cartilage luxé [12]. Ce traitement radical, bien que rarement proposé, semble avoir une efficacité proche de 100 % [13]. La place de la mésothérapie est parfois aussi discutée. Une meilleure connaissance du syndrome de Cyriax pourrait éviter nombre de retards diagnostiques, devant une douleur abdominale chronique.

Figure 5. Mécanisme du syndrome de Cyriax
Les douleurs abdominales en rapport avec une pathologie systémique à manifestation rhumatologique
Dans ce chapitre, ne seront bien entendu pas abordées les spondylarthropathies associées à des manifestations digestives de type maladies inflammatoires chroniques intestinales. Il s’agira essentiellement de traiter ici les douleurs abdominales observées au cours de certaines maladies inflammatoires comme le TRAP syndrome, la maladie périodique, le syndrome de Muckle-Wells, ou le syndrome Hyper-IgD (Table 2).
![Table 2. Caractéristiques principales et traits distinctifs de quatre fièvres périodiques héréditaires avec manifestations articulaires et abdominales intermittentes [42].](data:image/svg+xml;base64,PHN2ZyB4bWxucz0iaHR0cDovL3d3dy53My5vcmcvMjAwMC9zdmciIHdpZHRoPSI1NjAiIGhlaWdodD0iMzMwIiB2aWV3Qm94PSIwIDAgNTYwIDMzMCI+PHJlY3Qgd2lkdGg9IjEwMCUiIGhlaWdodD0iMTAwJSIgc3R5bGU9ImZpbGw6I2NmZDRkYjtmaWxsLW9wYWNpdHk6IDAuMTsiLz48L3N2Zz4=)
Table 2. Caractéristiques principales et traits distinctifs de quatre fièvres périodiques héréditaires avec manifestations articulaires et abdominales intermittentes [42].
TRAP syndrome
Le TRAPS (Tumor necrosis factor-Receptor-Associated Periodic Syndrome) est une fièvre héréditaire intermittente à transmission autosomique dominante liée à des mutations du récepteur 1A du TNF (le TNFRSF1A) se traduisant par des accès douloureux articulaires, abdominaux, musculaires, cutanés, oculaires. L’origine ethnique des patients est l’Europe du Nord, mais non exclusivement. En dehors des familles avec une transmission dominante, il existe des cas sporadiques. Sa prévalence est estimée entre 1 et 9 cas pour 1 million.
L’âge de début du TRAPS est variable, mais le plus souvent avant 20 ans. Les douleurs abdominales sont quasi constantes, les arthralgies, les myalgies fréquentes, les douleurs thoraciques possibles, de même que des céphalées, et chez les hommes des douleurs testiculaires. Les douleurs abdominales parfois extrêmement vives s’accompagnent le cas échéant de diarrhée ou de constipation, de nausées, et de vomissements. Les arthralgies présentes chez environ deux tiers des patients pendant les poussées sont monoarticulaires ou oligoarticulaires aux genoux, épaules, coudes, hanches ou encore aux doigts, poignets, articulations temporomandibulaires. Les arthrites sont plus rares ; un épanchement synovial, parfois responsable d’un flessum transitoire est possible. Les myalgies fréquentes touchent un groupe musculaire à la fois, volontiers à la racine d’un membre supérieur ou inférieur, avec gonflement, sensibilité, augmentation de la chaleur locale. Les douleurs thoraciques sont d’origine pleurale (pleurésie aseptique) ou musculaire pariétale. Une éruption cutanée est assez fréquente. La peau en regard des zones de myalgies est érythémateuse, avec un œdème prenant le godet. Les zones d’éruptions maculaires érythémateuses ou sous forme de plaques oedémateuses migratrices ne sont pas toutes siège de douleurs. L’éruption cutanée paraît épargner le visage. En revanche, un œdème péri-orbitaire unilatéral ou une conjonctivite douloureuse ou les deux ensemble sont possibles au cours des accès. Il n’y a pas de signes cutanés histologiques de vascularite, mais un aspect d’infiltrat périvasculaire et interstitiel de lymphocytes et de monocytes.
Un examen d’imagerie par résonance magnétique pratiqué dans le segment de membre symptomatique a montré dans un cas un aspect de myosite localisée avec anomalie du signal musculaire et du tissu sous-cutané sans atteinte des fascias ou des articulations adjacentes [14] et dans un autre cas [15] une fasciite confirmée à l’examen histologique (fasciite avec infiltrat de cellules mononucléées), sans atteinte musculaire.
L’évolution de l’accès douloureux est caractéristique. Les myalgies, l’érythème, l’atteinte articulaire progressent vers la région distale du membre en quelques jours ou semaines chez la majorité des patients (symptomatologie migratrice régionale évocatrice), avec régression sans séquelles. Les accès fébriles et douloureux transitoires durent rarement un à deux jours, souvent plus d’une semaine, et quelquefois plusieurs semaines.
Une réponse de phase aiguë est présente en crise avec une élévation de la C-réactive protéine, une hyperleucocytose à polynucléaires neutrophiles, et une consommation modérée du complément [16]. Les immunoglobulines sériques, en particulier les IgA sont possiblement augmentées. Quand il existe une élévation des IgD, elle reste en dessous de 100 UI/ml, contrairement au syndrome hyper-IgD.
Le dosage du TNFRSF1A soluble en inter-crise est inférieur à 1 ng/ml chez la plupart des patients ayant un TRAPS. Pendant un accès inflammatoire ses valeurs restent basses ou parfois deviennent transitoirement normales, alors que la concentration du TNF est augmentée. Le dosage du TNFRSF1A est interprété en fonction de la clairance de la créatinine. Il augmente en cas d’insuffisance rénale, par exemple si le patient a une localisation rénale d’amylose [17].
L’analyse de l’ADN génomique confirme typiquement une mutation du gène TNFRSF1A sur le chromosome 12p13. Les anomalies génétiques donnent des mutations faux-sens affectant les domaines extracellulaires du TNFRSF1A portant sur les cystéines qui sous-tendent sa structure spatiale [14, 18]. Le TRAPS est la situation où l’on a reconnu pour la première fois une mutation naturelle d’un récepteur du TNF. Dans certains cas, la symptomatologie est très évocatrice du TRAPS, mais on ne retrouve pas l’une des mutations actuellement attendues, ce que McDermott appelle le TRAPS négatif [19]. De tels patients doivent faire l’objet d’études nouvelles pour découvrir des mutations d’autres gènes liés ou non au TNFRSF1A [20].
La présence de mutations du TNFRSF1A est compatible avec le statut porteur sain (la symptomatologie pour les patients souffrant de TRAPS est de plus intermittente). Il est possible qu’il existe des formes de TRAPS à expression clinique fruste : fièvres récidivantes isolées, myalgies périodiques ou conjonctivites douloureuses périodiques, difficiles à rattacher au TRAPS [17].
Le nombre, l’intensité, la diversité, la durée des crises sont variables d’un sujet à l’autre et chez un même sujet d’une crise à l’autre. Les intervalles libres d’accès varient de quelques mois à plusieurs années, pendant lesquels le patient peut oublier sa maladie. Des facteurs déclenchent des accès : un traumatisme localisé, une infection bénigne. Tout se passe comme si la présence du TNFRSF1A muté est compatible avec l’absence de signes cliniques et il faut un facteur déclenchant pour provoquer les accès caractéristiques intermittents. Des complications peuvent résulter de la corticothérapie répétée ou d’autres traitements.
Une amylose AA, en particulier rénale, hépatique, menaçant le pronostic vital survient dans 25-40 % des cas de TRAPS. Elle semble varier selon les anomalies génétiques présentes dans les familles. Un sujet atteint d’amylose dans une famille implique une surveillance plus grande des autres membres de cette famille, mais le risque n’est pas absolu. Le dépistage se fait par le contrôle une fois par an ou tous les six mois de la protéinurie. Les dépôts d’amylose provenant de la protéine SAA (protéine amyloïde A sérique) sont visibles sur les coupes tissulaires colorées par le rouge Congo et prennent une coloration verte en lumière polarisée. Il est utile sur le long terme de contrôler les tests d’inflammation, quand le patient est en période sans manifestation clinique évidente, à cause du risque d’amylose AA.
La colchicine est inefficace et ne prévient pas les accès ; son action sur la prévention d’une amylose AA n’est pas démontrée dans le TRAPS. Les corticoïdes sont efficaces, à une dose orale supérieure à 20 mg par jour. Ils diminuent la sévérité et la durée des accès inflammatoires qui peuvent durer plusieurs semaines et dans certains cas devenir subintrants. La corticothérapie devient parfois moins efficace ou doit être prescrite plus fréquemment. L’hypothèse pathogénique d’un trouble de la libération membranaire du TNFRSF1A conduit à proposer un blocage de l’action du TNF. L’Etanercept, protéine de fusion comportant deux copies du récepteur p75 du TNF lié au fragment constant de l’Ig1 humaine est utilisée pour le traitement du TRAPS. La dose utilisée est de 25 mg d’Etanercept en injection sous-cutanée deux fois par semaine, mais pourrait être dans l’idéal adaptée selon le degré d’atteinte. Elle permet une atténuation des effets du TNF en se liant au TNF soluble et membranaire. Elle paraît intéressante, soit pour remplacer la corticothérapie, soit pour réduire les doses de corticoïdes pour contrôler les accès du TRAPS [21,22]. Dans une étude récente, elle ne permet pas d’éviter les accès [23].
Maladie Périodique (ou Fièvre Méditerranéenne Familiale (FMF)) [24, 25]
Les origines ethniques sont différentes de celles du TRAPS : juifs surtout sépharades, arméniens, arabes du Moyen-Orient, arabes maghrébins, turcs, et parfois autre origine méditerranéenne ou bien sujets n’étant ni d’origine méditerranéenne, ni juifs.
Comme au cours du TRAPS, les patients souffrent d’accès douloureux intermittents fébriles, articulaires, abdominaux, thoraciques (pleuraux), avec une réponse de phase aiguë, un risque d’amylose AA et chez les hommes de possibles épisodes de scrotite.
La transmission de la maladie périodique est en règle générale autosomique récessive à pénétrance incomplète, dite encore horizontale. La maladie périodique est retrouvée sur une seule génération, chez au moins un frère ou une sœur du patient. Néanmoins, même si les mutations s’expriment à l’état récessif dans la maladie périodique, la transmission dans certaines familles de maladie périodique peut apparaître « verticale » compte tenu de la grande fréquence de l’hétérozygotie dans les populations à risque. Sa prévalence est plus élevée que celle du TRAPS, entre 1 et 5/10 000.
L’anomalie génétique est différente entre la maladie périodique et le TRAPS. Le gène de la maladie périodique (sur le chromosome 16) appelé MEFV (ME pour mediterranean et FV pour fever) identifié en 1997, comprend dix exons, et code pour une protéine appelée pyrine ou marénostrine. Cette protéine fait partie de la famille des protéines RoRet, qui comprend aussi l’auto-antigène Ro52 et la RFP (Retfinger- protein). La pyrine/marénostrine s’exprime dans les polynucléaires neutrophiles. La recherche d’une des différentes mutations portant sur le gène MEFV est devenue un outil pour le diagnostic de la maladie périodique. D’autres gènes interviennent dans l’expression de la maladie, sa gravité, ou encore dans l’association avec une amylose AA [21,22].
La durée des crises différencie la maladie périodique et le TRAPS. Les crises dans la maladie périodique sont en règle générale plus courtes que dans le TRAPS. Dans la maladie périodique, la température s’élève à 38 ou 39, voire 40 °C en quelques heures, avec des frissons et se normalise en 12 à 36 heures, parfois quelques jours. Le sommet de la crise est atteint en deux à trois jours, et l’accès articulaire régresse en une semaine environ (de 3 jours à plusieurs semaines), sans habituellement de séquelle (font exception quelques cas avec une forme articulaire chronique en particulier à la hanche).
La fièvre disparaît avant la sédation des douleurs. Il n’existe ni œdème péri-orbitaire ni conjonctive douloureuse. Les myalgies localisées transitoires possibles dans la maladie périodique, sont toutefois plus évocatrices du TRAPS. L’éruption de la maladie périodique douloureuse siège parfois sur le tronc ou le visage, avec des placards cellulodermiques rouges vifs, mais surtout aux membres inférieurs, en dessous des genoux avec une peau rouge, infiltrée, ressemblant à un érysipèle, ou aux mains. Cette localisation cutanée aux mains ou pieds est évocatrice de la maladie périodique. Dans le TRAPS, l’éruption n’épargne que le visage.
Une péricardite est possible dans la maladie périodique, de même qu’une splénomégalie, une hépatomégalie (en dehors d’une localisation de l’amylose) et d’autres manifestations intermittentes, comme la méningite périodique aseptique de caractéristique discutée. Dans la maladie périodique un déficit de l’inhibiteur enzymatique du fragment C5a du système du complément est présent dans les liquides péritonéaux et synoviaux.
L’efficacité de la colchicine est en faveur de la maladie périodique. La colchicine à la posologie quotidienne de 1 mg en une ou deux prises (parfois 1,5 à 2 mg/jour) prévient les accès dans la majorité des cas et évite la constitution d’une amylose AA. La corticothérapie dans la maladie périodique pourrait réduire l’évolution de certains accès lorsqu’elle est utilisée précocement et à forte dose, mais avec un risque de rebond si elle est utilisée au long cours lors des essais de sevrage. En pratique la place de la corticothérapie au cours de la maladie périodique est habituellement très réduite.
Syndrome de Muckle-Wells
Le syndrome de Muckle-Wells et l’urticaire au froid familiale font aussi partie des fièvres héréditaires, avec comme pour le TRAPS, une transmission autosomique dominante. Le syndrome de Muckle-Wells décrit en 1962 dans une famille anglaise comporte en plus des arthralgies ou arthrites intermittentes avec une réponse de phase aiguë, une urticaire, une surdité de perception neurosensorielle d’aggravation progressive et un risque d’amylose, en particulier rénale. Les douleurs abdominales sont rares. Les anti- inflammatoires sont peu efficaces. L’anomalie génétique porte sur le gène CIAS1 du chromosome 1 (en 1q44) [26], tout comme pour l’urticaire au froid familiale [27]. Ce syndrome décrit en 1940 débute dans la petite enfance et se traduit par une urticaire survenant quelques heures après une exposition au froid, dans une série récente le délai minimal est de 90 minutes, et pour des températures inférieures ou égales à 22°C [28]. Il comporte aussi des accès douloureux, fébriles, avec frissons, arthralgies, arthrite, conjonctivite, et un risque d’amylose AA. La peau est parfois le siège aussi de macules, de plaques érythémateuses, de pétéchies. La protéine correspondante au gène CIAS1 a des régions homologues avec celle de la pyrine et est appelée compte tenu des circonstances de sa découverte, cryopyrine [29–31]. De plus en plus de familles sont décrites en Europe, et dans d’autres contrées. Ainsi au nord de l’Inde, plusieurs membres d’une famille ont une maladie caractérisée par un début précoce d’accès intermittents avec une fièvre durant au-delà d’une semaine, une urticaire, un œedème péri-orbitaire, des arthralgies modérées à sévères, une réponse de phase aiguë, une réponse à la colchicine, des cas d’amylose AA et des mutations sur le site chromosomique 1q44 [32].
Certains rapprochent également deux autres entités. Le CINCA (chronic, infantile, neurological, cutaneous and articular) syndrome appelé aussi NOMID (Neonatal infantile Onset Multisystem Inflammatory Disease), où des mutations sur le gène CIAS1 sont décrites [33,34]. Et la neutropénie cyclique appelée encore hématopoïèse cyclique autosomique dominante, caractérisée par des fluctuations cycliques du nombre de polynucléaires neutrophiles, de monocytes, d’éosinophiles, de lymphocytes, de plaquettes et de réticulocytes dans le sang circulant avec des cycles en moyenne de 21 jours, parfois 15 à 35 jours. Les périodes de neutropénie s’accompagnent de fièvre, de malaise, d’ulcération des muqueuses et parfois d’infections graves. Le gène de la neutropénie cyclique (ELA2) a été identifié et code pour une élastase des polynucléaires neutrophiles [35].
Le syndrome hyper-IgD
Le syndrome hyper-IgD identifié depuis 1983-1984 a des points communs avec le TRAPS : accès fébriles (non périodiques), arthralgies, arthrite, douleurs abdominales intenses fréquentes, avec souvent une diarrhée, des nausées, des vomissements. Les signes cliniques disparaissent progressivement en quelques jours, parfois plusieurs semaines. Les signes articulaires peuvent se traduire par une oligoarthrite, souvent symétrique, qui manque lors des premières crises. Une éruption maculaire ou papulaire est possible. Une réponse de phase aiguë est présente lors des accès. Le taux d’IgA sérique est souvent augmenté.
Beaucoup d’éléments opposent toutefois le syndrome hyper-IgD et le TRAPS. La plupart des patients souffrant du syndrome hyper-IgD sont originaires d’Europe mais surtout de Hollande, de France (toutefois non exclusivement). Le syndrome hyper-IgD débute en règle générale avant l’âge de dix ans, le plus souvent dès la première année (rarement chez l’adulte). L’examen retrouve des adénopathies, inguinales, axillaires et surtout cervicales, bilatérales, parfois une hépato-splénomégalie. Il n’y a pas d’accès pleuraux. Surtout le taux d’IgD est supérieur à 100 mU/ml ou encore 140 mg/L, à deux dosages à un mois d’intervalle. Il n’y a pas de risque d’amylose. La prédisposition au syndrome hyper-IgD est liée à des mutations (non constantes toutefois) du gène MVK codant la mévalonate kinase sur le chromosome 12q24 [36–38]. La mévalonate kinase intervient dans le métabolisme du mévalonate, intermédiaire dans la voie de synthèse du cholestérol. Le dosage de la mévalonate kinase lymphocytaire montre sa diminution [39]. Ce déficit enzymatique n’est pas aussi profond que dans l’acidurie mévalonique, qui est due à d’autres mutations du gène de la mévalonate kinase. L’efficacité de la colchicine et des anti- inflammatoires (AINS, corticothérapie) est inconstante et partielle [40]. Une réponse incomplète sous Etanercept a été notée [41].
Conclusion
Comme nous venons de le voir les étiologies rhumatologiques aux douleurs abdominales sont extrêmement nombreuses et variées. Il n’est d’ailleurs pas possible d’être parfaitement exhaustif, les causes allant d’origines mécaniques relativement simples à des tableaux systémiques inflammatoires très complexes.
Néanmoins il faut y penser lorsque les examens cliniques et paracliniques digestifs sont pauvres et après avoir donc éliminé une étiologie digestive, gynécologique, rénale ou vasculaire.
Un avis auprès d’un confrère rhumatologue permettra alors d’étayer le diagnostic et d’éviter ainsi parfois des explorations invasives inutiles.
Références
- Frexinos J, Denis P, Allemand H, Allouche S, Los F, Bonnelye G. Étude descriptive des symptômes fonctionnels dans la population générale française. Gastroenterol Clin Biol 1998;22:785-91.
- Ducrotte P. La douleur abdominale : le point de vue du gastro-entérologue. Gastroenterol Clin Biol 2003; 27 Sup.3;68-72.
- Vannimenus PY, Thevenon A, Duquesnoy B. Le dérangement intervertébral mineur dorso- lombaire : un syndrome souvent méconnu. NPN Médecine 1988;146:381-4.
- Sanou S, Belembaogo E, Nzenze JR, Moussavou Kombila JB, Bongo M, Boguikouma JB et al. Le syndrome de Cyriax : a` propos de 35 observations. Med Afrique Noire 1999;46.
- Monnin JL, Pierrugues R, Bories P, Michel H. Le syndrome de Cyriax, une cause d’erreur de diagnostic dans les douleurs abdominales. Press Med1989;17(1):25-9.
- Barki J, Blanc P, Michel J, Pageaux GP, Hacheman-aourag S, Carabalona JP et al. Painful rib syndrom (or Cyriax syndrome). Study of 100 patients. Press Med 1996;25(21):973-6.
- Udermann BE, Cavanaugh DG, Gibson MH, Doberstein ST, Mayer JM, Murray SR. Slipping Rib Syndrome in a collegiate swimmer: a case report. J Athl Train 2005;40(2):120-2.
- Scott EM, Scott BB. Painful rib syndrome – a review of 76 cases. Gut 1993;34:1006-8.
- Abbou S, Herman J. Slipping rib syndrome. Postgrad Med 1989;86(6):75-8.
- Meuwly JY, Wicky S, Schnyder P, Lepori D. Slipping Rib Syndrom, a place for sonography in the diagnosis of a frequently overlooked cause of abdominal or low thoracic pain. J Ultrasound Med 2002;21:339-43.
- Gregory PL, Biswas A, Batt ME. Musculoskeletal problems of the chest wall in athletes. Sports Med 2002;32:235-50.
- Copeland GP, Machin DG, Shennan JM. Surgical treatement of the ‘‘slipping rib syndrome’’. Br J Surg 1984;71:522-3.
- Machin DG, Shennan JM. Twelfth rib syndrome: a differential diagnosis ofloin pain. Br Med J 1983;287(6392):586.
- Dode C, Papo T, Fieschi C, Pecheux C, Dion E, Picard F, et al.A novel missense mutation (C30S) in the gene encoding tumor necrosis factor receptor 1 linked to autosomal-dominant recurrent fever with localized myositis in a French family. Arthritis Rheum 2000;43:1535–42.
- Hull KM, Wong K, Wood GM, Chu WS, Kastner DL. Monocytic fasciitis: a newly recognized clinical feature of tumor necrosis factor receptor dysfunction. Arthritis Rheum 2002;46:2189–94.
- McDermott EM, Smillie DM, Powell RJ. Clinical spectrum of familial Hibernian fever: a 14-year follow-up study of the index case and extended family. Mayo Clin Proc 1997;72:806–17.
- Drenth JP, van der Meer JW. Hereditary periodic fever. N Engl J Med 2001;345:1748–57.
- Galon J, Aksentijevich I, McDermott MF, O’Shea JJ, Kastner DL. TNFRSF1A mutations and auto-inflammatory syndromes. Curr Opin Immunol 2000;12:479–86.
- McDermott M. Fièvre récidivantes autosomiques dominantes. Aspects cliniques et génétiques. Rev Rhum [Ed Fr] 1999;66:555–63.
- Kriegel MA, Hüffmeier U, Scherb E, Scheidig C, Geiler T, Kalden JR, et al. Tumor Necrosis Factor-Receptor-Associated Periodic Syndrome characterized by a mutation affecting the cleavage site of the receptor: implication for pathogenesis. Arthritis Rheum 2003;48:2386–8.
- Nevala H, Karenko L, Stjernberg S, Raatikainen M, Suomalainen H, LagerstedtA, et al.A novel mutation in the third extracellular domain of the tumor necrosis factor receptor 1 in a Finnish family with autosomal- dominant recurrent fever. Arthritis Rheum 2002;46:1061–6.
- Drewe E, McDermott EM, Powell RJ. Treatment of the nephrotic syndrome with etanercept in patients with the Tumor Necrosis Factor- Receptor-Associated Periodic Syndrome. N Engl J Med 2000;343: 1044–5.
- Drewe E, McDermott EM, Powell PT, Isaacs JD, Powell RJ. Prospective study of anti-tumour necrosis factor receptor superfamily 1B fusion protein, and case study of anti-tumour necrosis factor receptor superfamily 1A fusion protein, in Tumor Necrosis Factor Receptorassociated- Periodic Syndrome (TRAPS): clinical and laboratory findings in a series of seven patients. Rheumatology (Oxford) 2003;42: 235–9.
- Samuels J, Aksentijevich I, Torosyan Y, Centola M, Deng Z, Sood R, et al. Familial mediterranean fever at the millenium. Clinical spectrum, ancient mutations, and a survey of 100 American referrals to the National Institutes of Health. Medicine (Baltimore) 1998;77: 268–77.
- Vinceneux P, Pouchot J, Méry JP.Maladie périodique (fièvre méditerranéenne familiale). 4e éd. In: Kahn MF, Peltier AP, Meyer O, Piette JC, editors. Maladies et syndromes systémiques, 1 vol. Paris: Médecine-Sciences Flammarion; 2000. p. 1137–268.
- Cuisset L, Drenth JP, Berthelot JM, Meyrier A, Vaudour G, Watts RA, et al. Genetic linkage of the Muckle-Wells syndrome to chromosome 1q44. Am J Hum Genet 1999;65:1054–9.
- Dode C, Le Du N, Cuisset L, Letourneur F, Berthelot JM, Vaudour G, et al. New mutations of CIAS1 that are responsible for Muckle-Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am J Hum Genet 2002;70:1498–506.
- Johnstone RF,DolenWK, Hoffman HM.Alarge kindred with familial cold auto-inflammatory syndrome. Ann Allergy Asthma Immunol 2003;90:233–7.
- Hoffman HM, Wright FA, Broide DH, Wanderer AA, Kolodner RD. Identification of a locus on chromosome 1q44 for familial cold urticaria. Am J Hum Genet 2000;65:1693–8.
- Hoffman HM, Mueller JL, Broide DH,Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold auto-inflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001;29:301–5.
- Hoffman HM, Wanderer AA, Broide DH. Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol 2001;108:615–20.
- Mc Dermott MF,Aganna E, Hitman GA, OgunkoladeBW, Booth DR, Hawkins PN. An autosomal dominant periodic fever associated with AA amyloidosis in a north Indian family maps to distal chromosome 1q. Arthritis Rheum 2000;43:2034–40.
- Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet 2002;71:198–203.
- Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal- onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated auto-inflammatory diseases. Arthritis Rheum 2002;46:3340–8.
- Horwitz M, Benson KF, Person RE, Aprikyan AG, Dale DC. Mutations in ELA2, encoding neutrophil elastase, define a 21-day biological clock in cyclic haematopoiesis. Nat Genet 1999;23:433–6.
- Drenth JP, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, de Jong JG, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper- IgD Study Group. Nat Genet 1999;22:178–81.
- Cuisset L, Drenth JP, Simon A, Vincent MF, van der Velde Visser S, van der Meer JW, et al. Molecular analysis of MVK mutations and enzymatic activity in hyper-IgD and periodic fever syndrome. Eur J Hum Genet 2001;9:260–6.
- Simon A, Cuisset L, Vincent MF, van Der Velde-Visser SD, Delpech M, van Der Meer JW, et al. Molecular analysis of the mevalonate kinase gene in a cohort of patients with the hyper-IgD and periodic fever syndrome: its application as a diagnosis tool. Ann Intern Med 2001;135:338–43.
- Frenkel J, Houten SM, Waterham HR, Wanders RJ, Rijkers GT, Kimpen JL, et al. Mevalonate kinase deficiency and Dutch type periodic fever. Clin Exp Rheumatol 2000;18:525–32.
- De Dios Garcia-Diaz J, Alvarez-Blanco MJ. Glucocorticoids but not NSAID abort attacks in hyper-IgD and periodic fever syndrome. J Rheumatol 2001;28:925–6.
- Arkwright PD, McDermott MF, Houten SM, Frenkel J, Waterham HR, Aganna E, et al. Hyper IgD syndrome (HIDS) associated with in vitro evidence of defective monocyte TNFRSF1A shedding and partial response to TNF receptor blockade with etanercept. Clin Exp Immunol 2002;130:484– 8.
- Masson et al. Le TRAPS. Revue du Rhumatisme 71 (2004) 565–572.
![Table I. Affections non digestives pouvant donner des douleurs à projection abdominale, le plus souvent aiguës. Référence [2]](https://www.fmcgastro.org/wp-content/uploads/2019/03/02_01_Table-1-560x225.jpg)

![Figure 4. Mécanisme d’une douleur pariétale par conflit du rameau antérieur d’un nerf cutané abdominal au niveau de sa gaine fibreuse au bord du muscle grand droit de l’abdomen [2]](https://www.fmcgastro.org/wp-content/uploads/2019/03/02_01_Figure-4-png.webp)

![Table 2. Caractéristiques principales et traits distinctifs de quatre fièvres périodiques héréditaires avec manifestations articulaires et abdominales intermittentes [42].](https://www.fmcgastro.org/wp-content/uploads/2019/03/02_01_Table-2-560x330.jpg)
