Liens d’intérêts
Conseiller pour Novartis, ITM, Terumo, Ipsen.
Dédommagement pour des interventions : Terumo, MSD, Esteve
Invitation à un congrès médical : Pierre Fabre et MSD
Mots-clés
Caractérisation initiale ; pluridisciplinarité ; réseau de soin.
Abréviations
5FU : 5-fluorouracile
ASS : analogue de la somatostatine
CAPTEM : capécitabine et témozolomide
DPC : duodéno-pancréatectomie céphalique
EI : effets indésirables
G1-3 : grade tumoral des TNE (de G1 à G3)
GTE : Groupe d’étude des Tumeurs Endocrines
IM : index mitotique
NEM1 : néoplasie endocrinienne multiple de type 1
MGMT : O6-méthylguanine-DNA méthyltransférase
MiNEN : tumeur mixte neuroendocrine-non neuroendocrine
OMS : organisation mondiale de la santé
RCP : réunion de concertation pluridisciplinaires
RIV : radiothérapie interne vectorisée
TNE-p : tumeur neuroendocrine pancréatique
TRO : taux de réponse objective
SG : survie globale
SSP : survie sans progression
STZ : streptozotocine
SZE : syndrome de Zollinger-Ellison
TAP : thoraco-abdomino-pelvien
TNCD : thésaurus national de cancérologie digestive
VHL : Von Hippel Lindau
VIP : vaso-intestinal peptide
Introduction et classification OMS
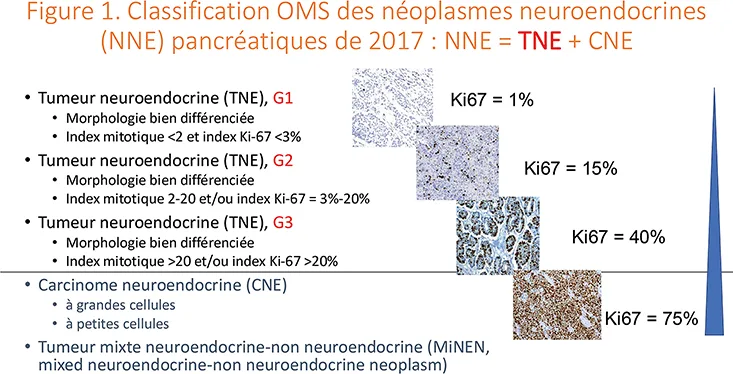
Selon la classification de l’OMS, les néoplasmes neuroendocrines sont séparés entre les carcinomes neuroendocrines, par définition peu différenciés, et les tumeurs neuroendocrines (TNE) qui sont bien différenciées (1). Les TNE sont ensuite classées en trois groupes selon le grade tumoral, qui prend en compte le nombre de mitoses (IM) /2 mm² et l’indice de prolifération Ki67 : NET-G1 (Ki67<3 % et IM<2), NET-G2 (Ki67 3-20 % ou IM 2-20), et NET-G3 (Ki67>20 % ou IM>20) (Figure 1).

Figure 1
Les tumeurs neuroendocrines pancréatiques (TNE-p) ont une incidence croissante mais restent des tumeurs rares [environ 1-2/100 000 habitants/an (2)]. Elles se forment à partir des ilôts endocrines du pancréas et peuvent produire des hormones (insuline, gastrine, glucagon, …). Avec les progrès de l’imagerie conventionnelle, la découverte fortuite de petite lésion pancréatique, asymptomatique (« incidentalome ») est de plus en plus fréquente. Cependant, les TNE-p restent fréquemment (~50 % des cas) avancées dès le diagnostic au niveau locorégional ou à distance. Elles ont une histoire naturelle bien plus favorable que les adénocarcinomes pancréatiques puisque même en présence de métastases à distance, le taux de survie à 5 ans dépasse les 50 %.
La prise en charge des TNE-p reste complexe car leur pronostic est très variable. Il est principalement influencé par l’index de prolifération Ki67 et le stade tumoral. Les dossiers doivent impérativement être discutés lors de réunions de concertations pluridisciplinaires (RCP) spécialisées, en France dans le cadre du réseau RENATEN-ENDOCAN (https://www.reseau-gte.org/renaten/). Les principaux objectifs thérapeutiques doivent être établis au cas par cas et partagés avec le patient à chaque étape de la prise en charge : guérison, augmentation de la durée de vie, contrôle local,
contrôle symptomatique, amélioration ou maintien de la qualité de vie, etc. Afin de guider cette prise en charge, la première étape est de parfaitement caractériser la TNE-p.
Importance de bien caractériser la tumeur neuroendocrine
On peut schématiser la caractérisation d’une TNE-p en 10 étapes indispensables (Figure 2).

Figure 2
- Peut-être encore plus que pour une tumeur plus agressive, il est essentiel d’avoir une idée claire des caractéristiques du patient, de son environnement et de ses projets de vie, puisque le plus souvent il n’est pas gêné par les symptômes de sa maladie (une fois pris en charge le syndrome sécrétoire s’il est présent), mais sa qualité de vie peut être plus altérée par les traitements prescrits.
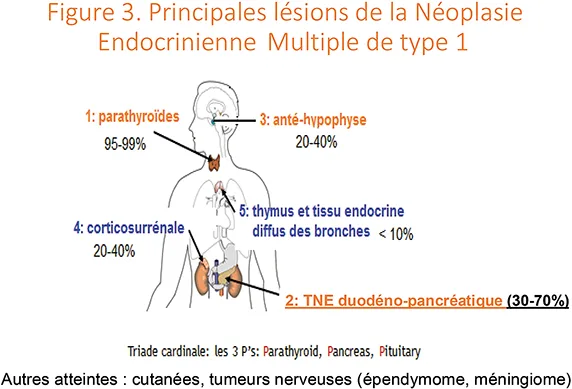
- Il faut aussi vérifier les antécédents personnels et familiaux pour éliminer un syndrome de prédisposition génétique, principalement la néoplasie endocrinienne multiple de type 1 (NEM1, Figure 3), qui va entraîner un dépistage dans la famille mais aussi modifier le type de chirurgie pancréatique en cas de TNE-p non métastatique.

Figure 3
Les TNE duodéno-pancréatiques liées à la NEM1 sont fréquemment multiples et fonctionnelles (environ 40 % des cas), le plus souvent des gastrinomes ou des insulinomes. À l’inverse, la probabilité de NEM1 est d’environ 25 % chez un patient présentant un gastrinome, et environ 5 % chez un patient présentant un insulinome, un VIPome ou un glucagonome. Les TNE-p non métastatiques associées à une NEM1 sont simplement surveillées lorsqu’elles sont asymptomatiques et ≤ 20 mm car associées à un faible risque de mortalité spécifique (3).
En cas de TNE-p métastatique, la présence d’une maladie de Von Hippel Lindau (VHL) peut permettre au patient de bénéficier du bezultifan, particulièrement efficace dans cette situation.
- Le diagnostic des TNE-p est souvent réalisé de manière fortuite, mais il faudra évaluer la présence de symptômes : i) soit liés aux masses tumorales comme toute tumeur pancréatique, de la tumeur primitive (ictère, douleur pancréatique, occlusion intestinale haute, ischémie par envahissement vasculaire) ou des métastases, et/ou ii) liés à un syndrome sécrétoire.
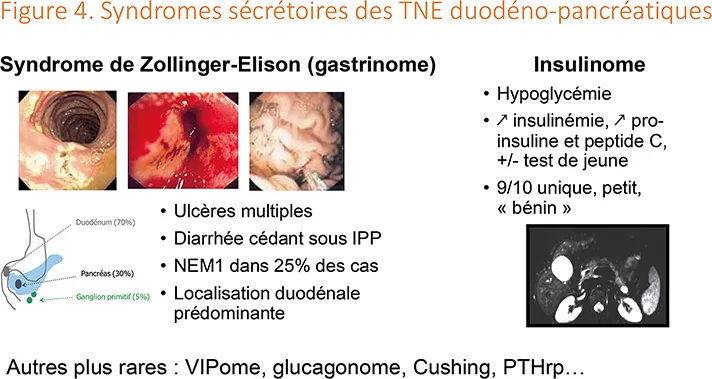
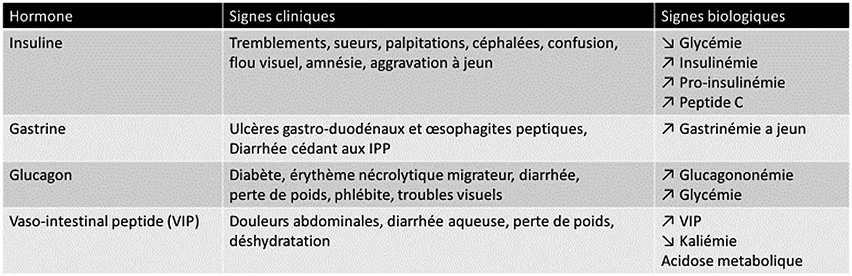
La recherche clinique d’un syndrome sécrétoire doit être systématique, et son traitement prioritaire, en raison du risque de complications parfois mortelles. L’indication du dosage des hormones et peptides endocrines doit être guidée par la clinique. Les deux plus fréquents sont le syndrome de Zollinger-Ellison (SZE, gastrinome) et l’insulinome (Figure 4 et Tableau 1) ; les autres syndromes sécrétoires sont bien plus rares (glucagonome, VIPome, cushing, PTH-rp, somatostatinome…).

Figure 4

Tableau 1 : Principaux syndromes sécrétoires associés aux TNE-p, adapté du TNCD (3)
Les insulinomes sont souvent de petites tumeurs de très faible risque évolutif. La principale complication est l’hypoglycémie et ses conséquences
(3). Si le diagnostic n’est pas évident, une épreuve de jeûne peut être réalisée en hospitalisation, idéalement dans un service d’endocrinologie.
Le syndrome de Zollinger-Ellison (SZE) résulte de l’hypersécrétion gastrique acide secondaire à une hypersécrétion de gastrine. Les gastrinomes sont généralement d’origine duodénale ou pancréatiques, et sont à haut risque de malignité. L’hypersécrétion acide gastrique peut occasionner des complications hydroélectrolytiques du fait de la diarrhée (déshydratation, hypokaliémie) et peptiques (œsophagite sévère, ulcères duodénaux pouvant se compliquer de perforation digestive ou d’hémorragie), potentiellement graves voire mortelles, à prévenir par IPP à doses suffisantes dès le diagnostic évoqué (3). Une diarrhée qui cède sous IPP est caractéristique d’un SZE. On verra souvent sur l’imagerie en coupe ou en écho-endoscopie un épaississement réactionnelle de la paroi gastrique (gros plis).
- À côté du dosage des hormones, de manière orientée par la clinique, on peut réaliser le dosage de la chromogranine A qui ne sera pas utile pour la prise en charge immédiate, mais qui peut être utilisé pour évaluer le pronostic initial et comme marqueur précoce de réponse d’une TNE-p métastatique par exemple. On peut cependant retenir que la chromogranine A n’est pas un bon marqueur tumoral, avec de nombreux faux positifs (dont les gastrites chroniques, la prise d’IPP….) et qu’on n’initiera jamais un traitement uniquement sur la seule évolution de son taux.
- Le plus important est probablement d’avoir une analyse histologique la plus précise et complète possible, dans le cadre du réseau ENDOCAN- PATH. Le pathologiste doit classer la TNE-p selon l’OMS (Figure 1), indiquer le grade tumoral et la valeur absolue de l’index de prolifération Ki67 (de 0 % à 100 %). En cas de TNE-p métastatique, il n’y a normalement pas besoin de réaliser une écho-endoscopie de la tumeur primitive, car il est préférable d’avoir alors le Ki67 sur la maladie métastatique souvent hépatique (moins invasif et plus informatif sur le pronostic), donc facilement accessible par une ponction biopsie hépatique. En cas d’indication de chimiothérapie, le pathologiste peut aussi déterminer le statut de la O6-méthylguanine-DNA méthyltransférase (MGMT) qui est à ce jour le seul biomarqueur prédictif de l’efficacité d’une chimiothérapie alkylante (meilleure réponse en cas de MGMT déficient) et pourrait faire préférer une chimiothérapie à base d’oxaliplatine ou un autre traitement systémique en cas de MGMT proficient (3).
Le bilan d’extension par des imageries morphologiques et nucléaire va guider la prise en charge initiale :
- Un scanner thoraco-abdomino-pelvien (TAP) avec injection triphasique pour mieux visualiser les TNE-p qui sont des tumeurs hypervasculaires est à réaliser en première L’IRM hépatique avec injection de gadolinium et séquences de diffusion sera plus sensible pour détecter de petites métastases hépatiques, celle-ci doit être systématique avant de discuter une chirurgie pancréatique.
- Le scanner TAP, l’IRM hépatique, +/- l’IRM du rachis (utile en cas de métastases osseuses pour évaluer le risque locorégional et pour le suivi thérapeutique) vont permettre avec l’imagerie nucléaire d’apprécier le volume tumorale (% envahissement hépatique et extra-hépatique) et ainsi appréhender où sera le pronostic de la maladie (tumeur primitive, foie, extra-hépatique).
- Une fois l’étendue de la maladie évaluée, le facteur pronostic le plus important (par définition non disponible à la première imagerie en coupe) est la pente évolutive des lésions. Il convient de l’évaluer au niveau de la TNE-p primitive et/ou des métastases hépatiques et/ou extra- hépatiques. Son évaluation doit se faire par deux imageries en coupe comparables (deux TDM TAP ou deux IRM). Une progression selon les critères RECIST en moins de 6 mois sera considérée par exemple comme rapide et en revanche, on sera conforté de continuer à surveiller une TNE-p centimétrique stable sur un an.
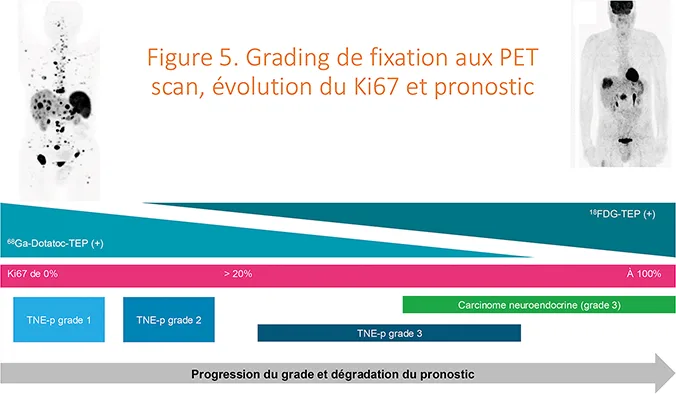
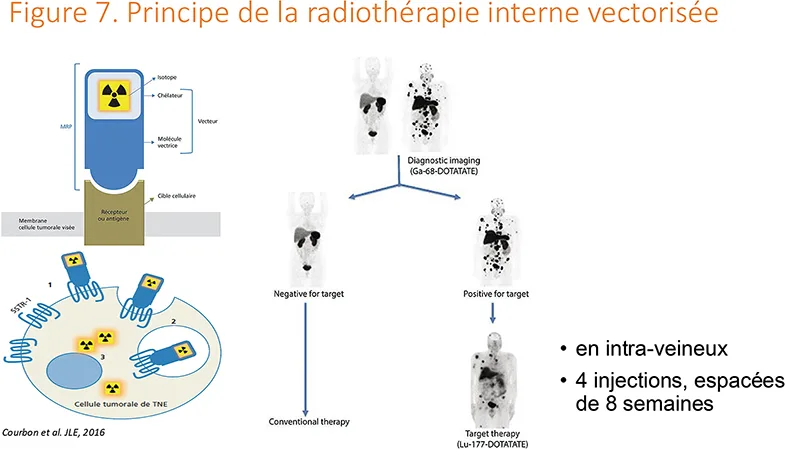
- L’imagerie nucléaire des récepteurs de la somatostatine par un TEP/TDM au 68Ga-Dotatoc est indispensable au bilan de toute TNE-p. Il va participer à parfaire le bilan d’extension (plus sensible sur la maladie ganglionnaire et osseuse), parfois trouver une tumeur primitive jusque-là non identifiée, et apprécier le volume tumoral global. Il existe des faux positifs (nodules de splénose, lymphome, granulomatose) à ce TEP puisque ces récepteurs peuvent être présents sur les lymphocytes. Il nous donne aussi des indications sur le pronostic de la TNE-p puisqu’une forte expression des récepteurs de la somatostatine est associée à un meilleur Ce sera le contraire pour le TEP/TDM au 18FDG qui sera d’autant plus fixant que la tumeur est proliférante (Figure 5). Enfin, il est un prérequis si on envisage une radiothérapie interne vectorisée (RIV) ciblant ces récepteurs. Le TEP/TDM au 18FDG présente donc un intérêt pronostique, mais il ne doit être réalisé que si son résultat est susceptible de faire changer la prise en charge.
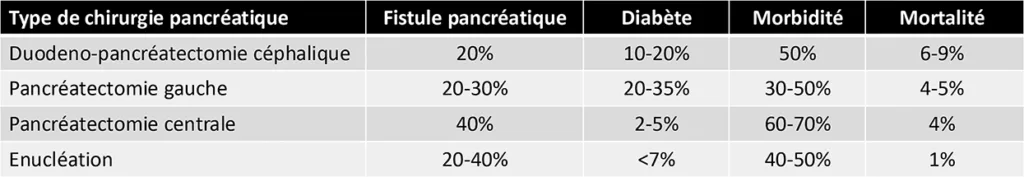
- Enfin, en cas de TNE-p non métastatique, l’IRM pancréatique et l’écho-endoscopie peuvent permettre de mieux localiser la/les TNE-p, en particulier les petits insulinomes parfois difficiles à Ils vont aussi participer à évaluer la résécabilité de la tumeur primitive (envahissement artériel péri-pancréatique, proximité avec le canal de Wirsung si une destruction par écho-endoscopie est envisagée, thrombose veineuse, circulation collatérale). Le type de chirurgie, fonction de la localisation de la tumeur pancréatique (tête, corps, queue), va forcément influencer la décision thérapeutique si une duodéno-pancréatectomie céphalique (DPC) est nécessaire, bien plus à risque de morbidité et mortalité qu’une pancréatectomie gauche (Tableau 2).

Figure 5
Tableau 2 : Principales complications post-opératoires selon le type de chirurgie pancréatique pour une TNE-p [adapté d’après (8)]
D’autant plus si on envisage une surveillance d’une TNE-p, l’enjeu majeur de la ponction sous écho-endoscopie est de permettre une analyse fiable des paramètres critiques pour l’évaluation de la malignité : différenciation, grade, index Ki67. Il existe une bonne corrélation entre l’expression du Ki67 déterminée à partir du matériel de ponction obtenu par écho-endoscopie et celui obtenu sur la pièce de résection chirurgicale, sous réserve d’un nombre de cellules suffisant. Dans l’étude de Larghi et al., il y avait une discordance significative dans 17 % (2/12) des cas entre la valeur du Ki67 déterminé sur le matériel de ponction et celui de la pièce de résection chirurgicale (4). L’écho-endoscopie avec contraste participe aussi à évaluer le pronostic, les tumeurs richement vascularisées ayant une moindre agressivité.
La prise en charge d’un syndrome sécrétoire est prioritaire
Le traitement des TNE-p présenté ici reprend largement ce qui est détaillé dans le TNCD (3). Le traitement antisécrétoire des TNE-p est une priorité et souvent une urgence, car les conséquences cliniques et biologiques de l’hypersécrétion hormonale sont source de morbidité et de mortalité. Le traitement par IPP doit être démarré immédiatement en cas de SZE sans attendre le résultat biologique de la gastrinémie. La dose d’IPP est adaptée à la réponse clinique et endoscopique, en débutant à 80-120 mg d’équivalent oméprazole. Les patients doivent être prévenus de la nécessité d’un traitement au long cours et de ne jamais l’interrompre. Concernant, l’insulinome, les mesures diététiques (fractionnement de l’alimentation et collations systématiques dans la journée) peuvent suffire en cas de syndrome sécrétoire peu intense. Le patient et son entourage sont informés des symptômes d’hypoglycémie et éduqués aux modalités de surveillance des glycémies capillaires et de resucrage. Le diazoxide (sulfamide hyperglycémiant inhibant la libération d’insuline) est efficace dans environ 50 % des cas, mais souvent pour une courte durée et il est mal toléré ; il est donné avant la résection/destruction de l’insulinome. Le traitement anti-sécrétoire des rares glucagonomes et VIPomes repose sur l’utilisation des formes retards des analogues de la somatostatine (octréotide ou lanréotide). Toutefois, en cas de syndrome sécrétoire non contrôlé de TNE-p, il faut rapidement envisager des traitements de réduction du volume tumoral (notamment hépatique) afin de contrôler les symptômes et diminuer le taux d’hormones circulantes, incluant potentiellement tous les traitements à visée antitumorale disponibles (embolisation intra-artérielle hépatique ou chirurgie de réduction tumorale, chimiothérapie, radiothérapie interne vectorisée, thérapies ciblées). Le pasiréotide (autre ASS) et l’évérolimus peuvent être utilisés pour leur effet proglycémiant contre l’insulinome métastatique. L’évérolimus est ainsi une option thérapeutique intéressante dès la première ligne contre les insulinomes métastatiques, non seulement parce que la survie sans progression (SSP) sous évérolimus est similaire à celle sous chimiothérapie (5), mais aussi en raison de son effet proglycémiant (6).
Traitement des TNE pancréatiques non métastatiques
Cas particulier des insulinomes
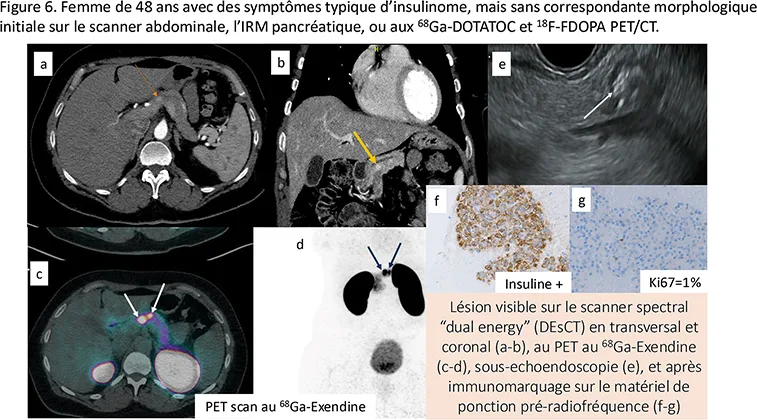
Le diagnostic d’insulinome peut être affirmé sur la présentation clinique et biologique sans retrouver de correspondance morphologique. Il convient alors de pousser les investigations avec des radiologues et écho-endoscopistes experts du pancréas puisqu’il existe des techniques sensibilisant la détection (exemple ci-dessus du scanner spectral « dual energy » avec coupes fines sur le pancréas, Figure 6a-b). Le TEP au 68Ga-Dotatoc n’est pas toujours contributif puisqu’environ 50 % des insulinomes n’ont pas de récepteurs de la somatostatine. En revanche, l’examen probablement le plus sensible pour identifier ces tumeurs, mais peu accessible en France (Paris et Lyon), est un PET au 68Ga-Exendine (Figure 6c-d).

Figure 6
La résection chirurgicale de l’insulinome est le traitement de référence des formes résécables. Une chirurgie limitée (sans curage systématique en cas de faible taille tumorale) est alors recommandée (par exemple, énucléation) lorsqu’elle est réalisable, étant donné le faible risque d’extension métastatique. La destruction par radiofréquence guidée par écho-endoscopie des insulinomes (Figure 6e) a montré une efficacité à court terme significative (>90 %) et peut être proposée en alternative à la chirurgie en première intention à condition qu’elle soit réalisée en centre expert puisque moins morbide que la chirurgie pancréatique (7). En cas de réponse clinique incomplète, une nouvelle séance de radiofréquence peut être envisagée et cela n’empêcherait pas une chirurgie pancréatique ultérieure. Ce type d’approche de destruction de la TNE-p, sans curage ganglionnaire associé, s’envisage pour les insulinomes car ces TNE-p sont dans plus de 90 % des cas de très bon pronostic (<2 cm, de grade 1, avec une hypervascularisation homogène, sans répercussion sur le canal de Wirsung) et n’ont donc pas besoin d’un traitement carcinologique.
Chirurgie de la TNE-p primitive
La résection chirurgicale doit toujours être discutée en RCP RENATEN. L’alternative d’une surveillance des « petits incidentalomes » de TNE-p prendra en compte l’âge et les comorbidités du patient, les caractéristiques de la TNE-p, le choix du patient et sa capacité à suivre une surveillance prolongée. À l’exception du cas particulier de l’insulinome décrit ci-dessus (énucléation ou destruction par radiofréquence, où l’épargne parenchymateuse est importante), l’indication d’une chirurgie se pose sur la nécessité d’un traitement carcinologique curatif. Ainsi, toute résection pancréatique doit être associée à un curage ganglionnaire avec un nombre minimal de 12 ganglions réséqués. Si une chirurgie d’épargne parenchymateuse (dont énucléation) est envisagée pour une TNE-p ayant des caractéristiques pronostiques favorables (taille ≤20 mm, G1, asymptomatique), par exemple dans un contexte de NEM1, elle doit être associée à la réalisation d’un picking ganglionnaire (3). Le type de geste de chirurgie pancréatique et ses risques associés (Tableau 2) sont à mettre en balance avec l’alternative d’une surveillance incluant le risque de laisser la maladie évoluer et de la traiter ensuite au stade métastatique (survie >5-10 ans pour une TNE-p métastatique de grade 1 métastatique).
L’indication de chirurgie carcinologique se pose dès que la TNE-p est de taille supérieure à 2 cm, ou symptomatique, ou avec suspicion de métastases ganglionnaires péri-pancréatiques, ou en présence de TNE-p progressive sur deux imageries en coupe identiques, et/ou avec des caractères morphologiques (pas de prise de contraste homogène, répercussion sur le canal de Wirsung) et histologiques (Ki67 élevé) de mauvais pronostic. Après une chirurgie pancréatique carcinologique, même en cas de résection R1, d’envahissement ganglionnaire, de Ki67 élevé, il n’y a pas de traitement adjuvant recommandé hors essais clinique (essai à venir dans cette situation, comparant une chimiothérapie adjuvante versus une surveillance).
En cas de NEM1, les indications chirurgicales sont identiques à celles des TNE-p sporadiques, à l’exception des gastrinomes pour lesquels le bénéfice de la chirurgie reste débattu en raison d’un rapport bénéfices/risques défavorable. La procédure chirurgicale doit être définie lors d’une RCP avec un chirurgien ayant une expérience de la prise en charge des patients atteints de NEM1 (exploration du duodénum par transillumination endoscopique et duodénotomie en cas de SZE, échographie per-opératoire pancréatique) et tenant compte des autres atteintes de la maladie et de son pronostic (9).
Prise en charge des TNE-p de type « Incidentalome »
Avec les progrès de l’imagerie conventionnelle, la découverte fortuite d’une TNE-p asymptomatique (« incidentalome ») est de plus en plus fréquente. La première étape est de confirmer le diagnostic de TNE-p et d’écarter un adénocarcinome. Ces TNE-p de petite taille (≤2 cm) asymptomatiques posent actuellement un problème de prise en charge, entre une chirurgie carcinologique non dénuée de risque pour des tumeurs le plus souvent d’extrêmement bon pronostic et une surveillance prolongée. L’objectif est de caractériser au mieux la TNE-p et évaluer son risque d’évolutivité (voir le chapitre ci-dessus). Des analyses moléculaires (DAXX/ATRX, signature « beta/alpha like ») sur le matériel histologique obtenue sous écho-endoscopie nous aideront probablement dans un avenir proche à encore mieux évaluer ce risque de malignité. Aujourd’hui, le TNCD (3) propose une surveillance des petits incidentalomes (<20 mm) de TNE-p sans résection chirurgicale si la découverte est fortuite et donc la tumeur est asymptomatique et non fonctionnelle ; la taille est <20 mm (stade T1) ; les caractéristiques sont typiques de TNE de bas grade avec une prise de contraste marquée à la phase artérielle en scanographie ou en IRM ou en échoendoscopie de contraste, hyperfixation caractéristique en TEP/ TDM au 68Ga-DOTATOC ; ils correspondent à des TNE-p de grade 1 (ou de faible grade 2 avec Ki67 <5 %, seuil non défini avec précision) si une preuve histologique a été obtenue (la biopsie sous EE est fortement recommandée en cas de taille >10 mm) ; il n’y a aucune suspicion de métastase ganglionnaire ou à distance ; il n’y a pas de dilatation canalaire pancréatique ou biliaire à l’imagerie ; il n’y a pas de progression sur les imageries de suivi ; le patient doit être compliant au suivi et avoir bien compris et consenti à l’option proposée. La surveillance des petits incidentalomes de TNE-p non réséqués peut être réalisée par IRM si elle permet la visualisation parfaite de la TNE, ou alternativement par scanographie (en tenant compte du risque à long terme d’irradiation cumulative). Bien qu’il ne soit pas précisément défini, un suivi peut être proposé à 6 mois puis annuellement. Les résultats préliminaires de 2 études prospectives observationnelles confortent cette prise en charge avec une surveillance active des patients ainsi sélectionnés, même si le suivi à long terme est pour l’instant limité. Dans l’étude Française IPANEM (10), 111 patients ont été inclus et seulement 3 (2,7 %) ont présenté des critères de malignité, 2 ont été opérés dans les 6 mois après inclusion (2 pN+ sur la pièce de résection chirurgicale, sans rechute à ce jour) et 1 patient non opéré a développé une maladie métastatique 12 mois après le diagnostic (la lésion était G1 de 15 mm). Aucun patient n’est pour l’instant décédé de sa TNE-p. Il en est de même dans l’étude européenne (11) qui a inclus 500 patients (406 dans le bras de surveillance active et 94 qui ont bénéficié d’une chirurgie) avec 3 décès non liés à la TNE-p après un suivi médian de 25 (interquartile 16-35) mois.
Bien que l’ablation par radiofréquence écho-endoscopie-guidée des petites TNE-p non fonctionnelles est techniquement faisable (12), son intérêt carcinologique (modifier l’histoire naturelle de la TNE-p, éviter une évolution métastatique, et in fine augmenter la survie) n’est pas démontré par rapport à une surveillance qui sera toujours moins à risque et plus médico-économiquement favorable. Elle n’est pas recommandée en routine hors essai clinique et son indication doit être validée en RCP RENATEN, notamment car elle ne permettra plus d’évaluer la pente évolutive de la TNE-p détruite (donc son risque de métastase ganglionnaire) sur lequel repose principalement l’indication chirurgicale ultérieure. Elle pourrait se poser en alternative par rapport à une surveillance active chez les patients non opérables ou trop comorbides, mais on peut aussi prendre le risque de laisser évoluer une TNE-p vers une maladie métastatique pour laquelle de nombreux traitements systémiques sont possibles et pour laquelle le pronostic au stade métastatique est en médiane supérieur à 5-10 ans.
Traitement des TNE pancréatiques métastatiques
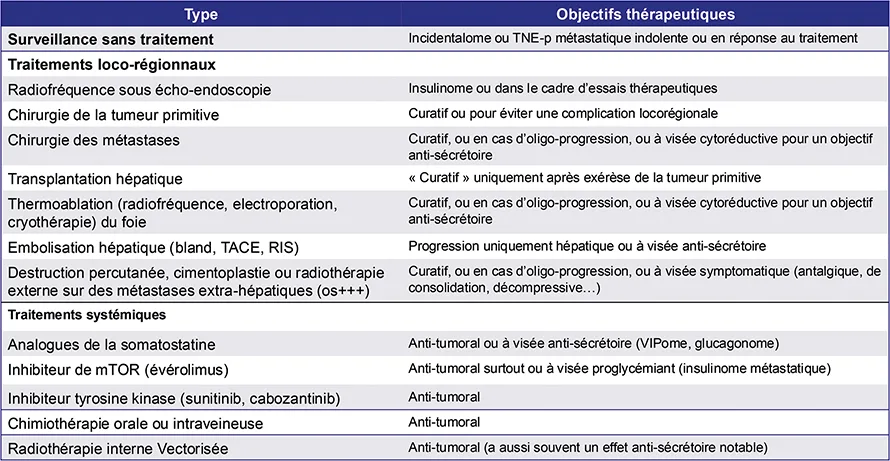
En cas de TNE-p métastatique, plusieurs traitements, systémiques ou locorégionaux, sont disponibles (Tableau 3). Ces derniers comprennent la résection chirurgicale de la tumeur primaire et/ou des métastases (curative ou palliative), les thérapies ablatives (type radiofréquence), l’embolisation du foie (seule ou associée à la chimiothérapie) et la radiothérapie interne sélective du foie (non remboursée en France dans cette situation). Parmi les thérapies systémiques pour les TNE-p, deux types de traitements ciblant les récepteurs de la somatostatine peuvent être prescrits, comprenant deux analogues de la somatostatine [ASS, lanréotide (13) et octréotide (14)] et une radiothérapie interne vectorisée [RIV, 177Lu-edotréotide (15), mais accessible dans un certain cadre, Figure 7], deux thérapies ciblées comprenant un inhibiteur de mTOR [évérolimus (16)] et deux inhibiteurs de la tyrosine kinase [sunitinib approuvé pour les TNE-p (17) et probablement bientôt le cabozantinib (18)], et deux types de chimiothérapies cytotoxiques comprenant un agent alkylant (tel que la streptozotocine (19), et le temozolomide (20) ou la dacarbazine) et une chimiothérapie à base d’oxaliplatine (21).

Tableau 3

Figure 7
La rareté et l’hétérogénéité des TNE-p expliquent le faible nombre d’études randomisées et le faible niveau de preuve associé. Promouvoir les inclusions des patients dans les essais thérapeutiques doit être une priorité, même dans les situations où il existe des recommandations thérapeutiques. Une liste des études en cours, régulièrement mise à jour, est disponible sur le site du Groupe d’étude des Tumeurs Endocrines (GTE) (https://www.reseau-gte.org/protocoles-du-gte).
Métastases non ou peu progressives, macroscopiquement résécables
Bien qu’il n’y ait pas de définition validée, une progression lente peut être arbitrairement définie par une augmentation de la taille de la tumeur ≤20 % (critères RECIST) en 12 mois. La résection et/ou la destruction (percutanée ou peropératoire) de toutes les métastases visibles doit toujours être discutée, dans la mesure du possible (en combinant éventuellement différentes procédures et différentes modalités et en prenant en compte les risques de complications associées) (3, 22). En effet, cette stratégie semble être associée à la survie la plus prolongée, bien qu’elle n’ait jamais été correctement comparée à d’autres traitements. Néanmoins, la récidive est généralement la règle en raison de tumeurs microscopiques persistantes chez tous les patients et ce type de prise en charge agressive ne doit se discuter qu’après avoir vérifié que l’histoire naturelle est favorable (3). Les chirurgies hépatiques en deux étapes, ou une hépatectomie mineure associée à une destruction des métastases des autres segments par radiofréquence peropératoire doivent être mises en balance avec le risque presque inévitable de récidive métastatique à long terme, la morbidité et la mortalité des procédures extensives, et le volume hépatique restant qui pourrait limiter l’administration ultérieure de traitements loco-régionaux ou à distance.
Métastases non résécables
La résection de la TNE-p primitive en cas de métastases peut se discuter en RCP RENATEN, surtout si le geste n’est pas une DPC, afin d’éviter d’éventuelles complications locales (hypertension portale) ou d’anticiper une transplantation hépatique, mais uniquement après plusieurs mois de surveillance ou de traitement systémique. La transplantation hépatique se discute dans des cas exceptionnels (<10 transplantations hépatiques par an en France dans cette indication) en cas de métastases hépatiques diffuses non résécables, non ou très lentement évolutives, avec un Ki67 bas (<5-10 %, seuil non déterminé précisément), en l’absence de métastases extra-hépatiques, chez un patient jeune (<55-60 ans), avec une tumeur primitive déjà réséquée et après un recul évolutif suffisant à partir du diagnostic de la maladie métastatique (3).
Au départ, les médecins doivent concevoir une séquence thérapeutique complète a priori, dont chaque étape doit être rediscutée en fonction des résultats du traitement précédent. Plusieurs questions doivent être discutées à chaque étape : si le pronostic et les symptômes sont liés à la tumeur primitive ou aux métastases, au volume tumorale et/ou à la sécrétion hormonale. La nécessité de réduire rapidement la charge tumorale (en raison des symptômes liés à la tumeur et du risque de complications locales) ou d’arrêter une progression rapide sera plus souvent satisfaite par un traitement locorégional (lorsque cela est possible, chirurgie ou traitement radiologique), la chimiothérapie ou la RIV. En revanche, si l’objectif du traitement est de prolonger la qualité de vie d’un patient asymptomatique présentant de bons facteurs pronostiques, il convient de cibler les traitements présentant le meilleur profil de sécurité, comme les ASS (13). Le choix d’un traitement est fonction de trois objectifs différents : viser la plus grande efficacité du traitement, viser le meilleur profil de sécurité, ou suivre les préférences du patient. Aucun de ces objectifs n’étant meilleur que les autres en soi, le contexte particulier de chaque situation doit être pris en compte.
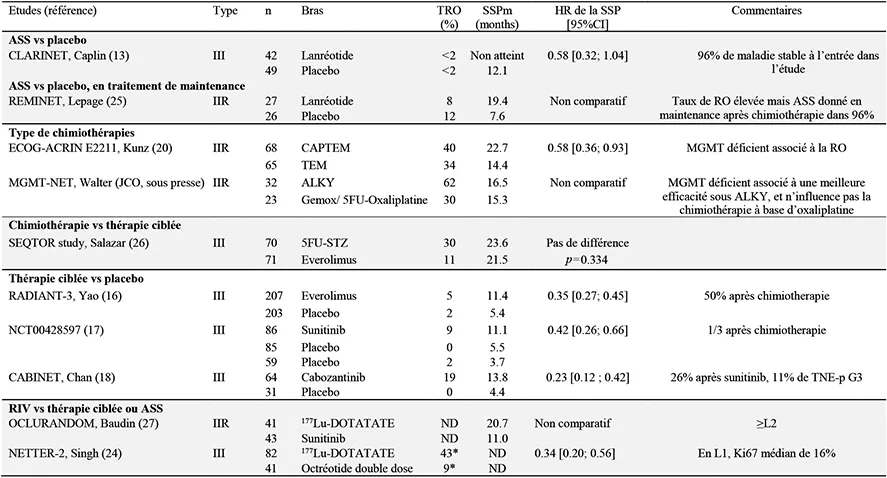
Concernant l’efficacité, si on regarde les résultats des essais cliniques randomisés récents, qui utilisent la survie sans progression (SSP) et le taux de réponse objective (TRO) comme principaux critères de jugement (Tableau 4), on peut dire qu’en terme d’efficacité : les ASS sont associés à une amélioration de la SSP par rapport à une stratégie d’observation (placebo) pour les TNE-p (13) ; la double chimiothérapie (CAPTEM, capécitabine et témozolomide) est associée à une amélioration de la SSP et du TRO par rapport au témozolomide (TEM) dans les TNE-p, même si les résultats sont déjà bons pour le TEM en monothérapie chez les patients qui ne tolèreraient pas la capécitabine (CAP) (20) ; la double chimiothérapie (5FU-Streptozotocine) est associée à une SSP similaire à celle de l’évérolimus dans les TNE-p, mais avec un TRO plus élevé sous chimiothérapie (5) ; l’évérolimus (16), le sunitinib (17) et le cabozantinib (18) sont associés à une amélioration de la SSP et du TRO par rapport au placebo dans les TNE-p ; même si l’essai n’était pas comparatif, la SSP est numériquement plus longue sous RIV (177Lu) qu’avec le sunitinib (23). De plus, la RIV est associée à une amélioration de la SSP et du TRO par rapport à la double dose d’ASS en première ligne de traitement d’une TNE-p (54 % de primitif pancréatique dans cette étude) de grade 2-3 (Ki67 entre 10 % et 55 %) (24). Cependant, il faut attendre les résultats de COMPOSE (NCT04919226), pour savoir si la RIV est plus efficace que la chimiothérapie, qui est le traitement de référence dans cette situation.

Tableau 4
Si l’on prend la tolérance/sécurité thérapeutique comme objectif principal de notre traitement (ne pas dégrader la qualité de vie d’un patient asymptomatique), il convient de différencier la toxicité à court et long terme. La toxicité à court terme, c’est-à-dire la tolérance au traitement, est estimée par la fréquence et le grade des effets indésirables (EI). Certains EI peuvent être très fréquents, mais de faible grade, et avoir néanmoins un impact sur la qualité de vie du patient. Cet aspect doit être considéré en tenant compte de la durée du traitement (par exemple, fréquence élevée d’EI pour l’embolisation du foie ou la chimiothérapie IV, mais ces traitements sont administrés pendant une courte période). La fréquence des EI symptomatiques, leur degré et la durée du traitement auront un impact sur la qualité de vie et les préférences des patients. Concernant le risque de toxicité à long terme, c’est-à-dire de séquelles, celles-ci sont généralement plus rares mais importantes, car elles peuvent empêcher l’administration d’autres traitements. En ce qui concerne les traitements systémiques, les ASS sont les seuls pour lesquels il n’y a pas de risque cumulatif, et dont l’administration à très long terme peut être effectuée sans risque de séquelles. Les risques de lithiase biliaire et d’insuffisance exocrine pancréatique sont facilement gérables en pratique courante. En revanche, les chimiothérapies, les thérapies ciblées (évérolimus, sunitinib et cabozantinib), ou la RIV nécessitent de la prudence, un suivi clinique et biologique régulier car les EI sont plus fréquents sous traitement, mettent parfois en jeu le pronostic vital (de grade 3-4), et/ou sont associés à un risque de séquelles telles que l’insuffisance rénale (streptozotocine, thérapies ciblées, et RIV), la cardiotoxicité (sunitinib et cabozantinib), et la toxicité hématologique (tous sauf les ASS). Le choix entre une double chimiothérapie (CAPTEM) et une monothérapie par temozolomide (TEM) dans les TNE-p doit tenir compte des toxicités (doublement des taux de toxicité de grade 3-4 dans le bras CAPTEM par rapport au bras temozolomide), contrebalancées par l’efficacité supérieur du CAPTEM. Le choix entre l’évérolimus et le sunitinib, qui semblent avoir une efficacité relativement similaire dans les TNE-p, sera principalement déterminé par leurs profils respectifs de contre-indication et de toxicité, qui sont plus métaboliques pour l’évérolimus et cardiovasculaires pour le sunitinib/cabozantinib. Un autre point très important, qui n’est pas étudié dans les essais cliniques randomisés est le risque cumulatif de l’administration de ces différents traitements en association (rarement) ou séquentiellement. Berdelou et al. ont rapporté que sur une durée médiane de SG de 5 ans, la fréquence des événements toxiques chroniques augmentait avec le nombre de lignes reçues, de 4 % après la première ligne à 24 % après cinq lignes (25). Deux exemples classiques de ces toxicités chroniques concernent l’insuffisance rénale chronique sévère qui peut apparaître sur 3-5 ans en raison de la néphrotoxicité cumulée de chaque traitement (steptozotocine, évérolimus, sunitinib, RIV, iode en raison des TDM multiples ou lors de l’embolisation hépatique) et le risque de syndrome de myélodysplasie/leucémie aiguë, qui peut être augmenté par l’association d’agents alkylants et de RIV (26). Ceci illustre l’importance des données en vie réelle pour l’évaluation du risque de toxicité cumulative, ce qui est l’objectif de l’étude TOLERATE qui va se mettre en place courant 2025. Enfin, l’ablation chirurgicale de la tumeur primitive, est risquée en post-opératoire bien sûr, mais aussi en termes d’effets à long terme comme l’insuffisance pancréatique exocrine après résection duodéno-pancréatique qui constituera aussi une contre-indication relative à des embolisations hépatiques du fait d’un risque accru de complications infectieuses.
En somme, si l’on ne considère que les traitements systémiques disponibles, il existe au moins 6 options pour les TNE-p (Tableau 3). En théorie, 3 options thérapeutiques correspondent à 6 séquences possibles, 4 options thérapeutiques à 24 séquences, 5 options thérapeutiques à 120 séquences, etc. Heureusement, des recommandations existent, principalement guidées par la présence de symptômes, le volume tumoral, le grade de la tumeur/ pente évolutive et l’expression des récepteurs de la somatostatine (3, 22).
Les ASS sont le traitement initial recommandé pour les TNE-p métastatique de grade 1-2 (Ki67<10 %), et/ou de faible volume tumorale, ou à croissance lente. En revanche, la chimiothérapie (à base de streptozotocine ou de témozolomide) est considérée comme le traitement systémique standard d’emblée pour les patients présentant une maladie volumineuse, une croissance tumorale rapide (progression RECIST sur moins d’un an), un Ki67 élevé (>10 %) et/ou des TNE-p symptomatiques. Lorsque le traitement vise à réduire la charge tumorale rapidement, une bi-chimiothérapie (CAPTEM ou 5FU-STZ) est préférable. Pour les patients chez qui une réponse objective n’est pas nécessaire, une thérapie ciblée pourrait être une option de traitement de première ligne (comme l’a montré l’étude SEQTOR avec l’évérolimus) (5).
En deuxième ligne, après un ASS ou une chimiothérapie, l’évérolimus et le sunitinib sont recommandés dans les TNE-p progressives de type G1-2, tandis que le belzutifan est le traitement de choix pour les patients atteints de TNE-p avancées liées à VHL (3). Les patients atteints d’une TNE-p avec expression des récepteurs de la somatostatine peuvent être traités par RIV avant l’administration de thérapies ciblées (23, 24). Son accessibilité est néanmoins actuellement réduite en France dans cette indication en raison de l’absence de remboursement spécifique.
Conclusions
La prise en charge des TNE-p reste complexe car leur pronostic est très variable. La prise en charge d’un syndrome sécrétoire (principalement insulinome et SZE) est prioritaire. De nombreux traitements sont disponibles, qu’ils soient loco-régionaux (dont la chirurgie, la radiofréquence, et l’embolisation hépatique) ou systémiques (cinq classes thérapeutiques dont la RIV). Les dossiers doivent impérativement être discutés, à chaque étape, lors de RCP dédiées dans le cadre du réseau RENATEN-ENDOCAN. Promouvoir les inclusions des patients dans les essais thérapeutiques doit être une priorité dans cette maladie rare et complexe.
Références
- WHO classification of Tumours Editorial Board; Digestive System Tumours, WHO Classification of Tumours, 5th Edition. 2019;12019.
- White BE, Rous B, Chandrakumaran K, Wong K, Bouvier C, Van Hemelrijck M, et Incidence and survival of neuroendocrine neoplasia in England 1995-2018: A retrospective, population-based study. Lancet Reg Health Eur. 2022;23:100510.
- de Mestier L WT, Hadoux S, Cros J, Deguelte S, Gaujoux S, Hautefeuille V, et al. « Néoplasies Neuroendocrines Digestives ». Thésaurus National de Cancérologie Digestive, Novembre 2023, [http://www.tncd.org].
- Larghi A, Capurso G, Carnuccio A, Ricci R, Alfieri S, Galasso D, et Ki-67 grading of nonfunctioning pancreatic neuroendocrine tumors on histologic samples obtained by EUS-guided fine-needle tissue acquisition: a prospective study. Gastrointest Endosc. 2012;76(3):570-7.
- Salazar R TSKMTAG-CRKHJCBSIEBTEMJR, editor Randomized open label phase III study comparing the efficacy and safety of everolimus followed by chemotherapy (CT) with streptozotocin (STZ)-5FU upon progression or the reverse sequence, in advanced progressive panNETs: The SEQTOR study (GETNE 1206). ESMO; 2022; Paris.
- Bernard V, Lombard-Bohas C, Taquet MC, Caroli-Bosc FX, Ruszniewski P, Niccoli P, et al. Efficacy of everolimus in patients with metastatic insulinoma and refractory hypoglycemia. Eur J Endocrinol. 2013;168(5):665-74.
- Crinò SF, Napoleon B, Facciorusso A, Lakhtakia S, Borbath I, Caillol F, et al. Endoscopic Ultrasound-guided Radiofrequency Ablation Versus Surgical Resection for Treatment of Pancreatic Insulinoma. Clin Gastroenterol Hepatol. 2023;21(11):2834-43.e2.
- Tamburrino D, Partelli S, Renzi C, Crippa S, Muffatti F, Perali C, et Systematic review and meta-analysis on laparoscopic pancreatic resections for neuroendocrine neoplasms (PNENs). Expert Rev Gastroenterol Hepatol. 2017;11(1):65-73.
- Gaujoux S, Martin GL, Mirallié E, Regenet N, Le Bras M, Pattou F, et al. Life expectancy and likelihood of surgery in multiple endocrine neoplasia type 1: AFCE and GTE cohort study. Br J Surg. 2022;109:872-9.
- Gincul R NB, Perrier M, Karsenti D, Palazzo L, Maire F, Borbath I, et al. editor Incidentalomes pancréatiques neuroendocrines non fonctionnels de petite taille (≤ 2 cm) : quel risque évolutif à 3 ans ? Résultats d’une étude prospective multicentrique nationale JFHOD; 2023; Paris.
- Partelli S, Massironi S, Zerbi A, Niccoli P, Kwon W, Landoni L, et al. Management of asymptomatic sporadic non-functioning pancreatic neuroendocrine neoplasms no larger than 2 cm: interim analysis of prospective ASPEN trial. Br J Surg. 2022;109(12):1186-90.
- Barthet M, Giovannini M, Gasmi M, Lesavre N, Boustière C, Napoleon B, et Long-term outcome after EUS-guided radiofrequency ablation: Prospective results in pancreatic neuroendocrine tumors and pancreatic cystic neoplasms. Endosc Int Open. 2021;9(8):E1178-e85.
- Caplin ME, Pavel M, Cwikla JB, Phan AT, Raderer M, Sedlackova E, et Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-33.
- Rinke A, Muller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, et Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-63.
- Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, et Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N Engl J Med. 2017;376(2):125-35.
- Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, et Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-23.
- Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, et Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501-13.
- Chan JA, Geyer S, Zemla T, Knopp MV, Behr S, Pulsipher S, et al. Phase 3 Trial of Cabozantinib to Treat Advanced Neuroendocrine N Engl J Med. 2024.
- Moertel CG, Lefkopoulo M, Lipsitz S, Hahn RG, Klaassen Streptozocin-doxorubicin, streptozocin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1992;326(8):519-23.
- Kunz PL, Graham NT, Catalano PJ, Nimeiri HS, Fisher GA, Longacre TA, et A Randomized Study of Temozolomide or Temozolomide and Capecitabine in Patients with Advanced Pancreatic Neuroendocrine Tumors (ECOG-ACRIN E2211). J Clin Oncol. 2022:101200JCO2201013.
- Girot P, Baudin E, Senellart H, Bouarioua N, Hentic O, Guimbaud R, et al. Oxaliplatin and 5-Fluorouracil in Advanced Well-Differentiated Digestive Neuroendocrine Tumors: A Multicenter National Retrospective Study from the French Group of Endocrine Tumors. Neuroendocrinology. 2022;112(6):537-46.
- Pavel M, Oberg K, Falconi M, Krenning EP, Sundin A, Perren A, et al. Gastroenteropancreatic neuroendocrine neoplasms: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31(7):844-60.
- Baudin E WTBASDHJLCTDACDLDMLDEQE, for the E-Rn, Gte, editors. First multicentric randomized phase II trial investigating the antitumor efficacy of peptide receptor radionucleide therapy with 177lutetium–octreotate (OCLU) in unresectable progressive neuroendocrine pancreatic tumor: results of the OCLURANDOM trial. ESMO; 2022; Paris.
- Singh S HD, Myrehaug S, et al. [177Lu]Lu-DOTA-TATE plus longacting octreotide versus highdose long-acting octreotide for the treatment of newly diagnosed, advanced grade 2–3, well-differentiated, gastroenteropancreatic neuroendocrine tumours (NETTER-2): an openlabel, randomised, phase 3 study. Lancet. 2024.
- Berdelou A, Boige V, Arfi-Rouche J, Malka D, Ederhy S, Izzedine H, et al. All Patients with a Pancreatic Neuroendocrine Tumour (pNET) Will not Benefit from all Approved or Recommended Therapeutic Options: A Real Life Retrospective Study. 2016.
- Brieau B, Hentic O, Lebtahi R, Palazzo M, Ben Reguiga M, Rebours V, et al. High risk of myelodysplastic syndrome and acute myeloid leukemia after 177Lu-octreotate PRRT in NET patients heavily pretreated with alkylating Endocr Relat Cancer. 2016;23(5):L17-23.