LIENS D’INTÉRÊT
Gilead (Orateur), Novartis (Expertise), La Jolla (Expertise), Intercept (Orateur).
MOTS-CLÉS
Ferritine, Hémochromatose, Syndrome métabolique, Maladie rares.
Introduction
L’hyperferritinémie est un motif très fréquent de consultation en hépato-gastro-entérologie. Si la prise en charge dans le cadre d’une hémochromatose HFE C282Y est bien codifiée, l’hyperferritinémie ne relève pas de ce cadre dans la majorité des cas et la conduite à tenir est alors moins clairement établie.
Il est fondamental de rappeler que la ferritine est une protéine intracellulaire de stockage du fer. Le taux sérique (ferritinémie) est donc un reflet indirect du stock en fer. De ce fait, et parce que le fer est un élément impliqué dans plusieurs mécanismes physiologiques, de très nombreuses causes peuvent induire une hyperferritinémie, qu’elle soit ou non associée à une réelle surcharge en fer.
La bonne connaissance et compréhension de ces facteurs est indispensable à la prise en charge adéquate d’une hyperferritinémie. Il faudra dans un premier temps analyser les facteurs présents pouvant influer le taux de ferritine pour ne pas s’orienter à tort vers une pathologie du métabolisme du fer. Dans certaines situations la quantification de la concentration hépatique en fer permet de conforter l’orientation et potentiellement d’évoquer une surcharge d’origine génétique autre que l’hémochromatose classique. Seul un diagnostic correct permettra de décider si une prise en charge thérapeutique spécifique est nécessaire.
Étiologies
Les causes d’hyperferritinémie
Ferritine et syndrome métabolique
L’augmentation de la prévalence du syndrome métabolique (1) et l’intrication étroite des métabolismes des lipides, des glucides et du fer (2-4) font du syndrome métabolique la première cause de consultation pour hyperferritinémie.
La ferritine marqueur prédictif du syndrome métabolique
Bien que les relations entre ferritine et insulino-résistance ne soient pas clairement élucidées, des études ont bien démontré le caractère prédictif de l’hyperferritinémie. Dans une cohorte prospective de 13 084 hommes indemnes de syndrome métabolique et suivis pendant 5 ans, Park et al. ont démontré, en ajustant pour les cofacteurs connus, que l’hyperferritinémie était associée à l’apparition d’un syndrome métabolique au cours du suivi (5). De façon similaire dans une cohorte européenne cherchant à identifier les facteurs de risque d’apparition d’un diabète de type 2 (340 234 individus suivis pendant 11 ans), l’hyperferritinémie (ajustée sur les autres facteurs de risques connus) était proportionnellement associée au risque de développer un diabète de type 2 (6).
Il peut donc exister une hyperferritinémie d’origine métabolique, avant même l’apparition clinique du syndrome métabolique. Par ailleurs l’hyperferritinémie est associée à une augmentation du risque de mortalité cardiovasculaire (7) et hépatique (8).
La ferritine marqueur de sévérité de la maladie stéatosique du foie
Une fois le syndrome métabolique constitué l’hyperferritnémie est fréquente, on la trouve par exemple chez 17,8 à 29,7 % des patients intolérants au glucose et 35 % des patients diabétiques (9). Son augmentation est proportionnelle à l’insulino- résistance (10, 11).
Dans le cadre de la maladie stéatosique du foie l’hyperferritinémie est présente chez 20 % des patients (12) dont 35 à 45 % auront une surcharge en fer associée au plan histologique (12, 13). C’est une surcharge dont la répartition peut être macrophagique, mixte, ou hépatocytaire sans que les mécanismes physiologiques sous-tendant cette surcharge soient bien compris (14). La présence d’une surcharge en fer est associée de façon indépendante à un stade de fibrose plus sévère (13, 15), et l’hyperferritinémie à un score NAS (NAFLD Activity Score) plus élevé sur toutes ses composantes (stéatose, inflammation, ballonisation) (12).
Syndrome métabolique et surcharge en fer secondaire : l’hépatosidérose métabolique
L’hépatosidérose métabolique se définit par l’association d’une surcharge en fer inexpliquée et d’un contexte métabolique associant obésité abdominale, et/ou hypertension artérielle, et/ou dyslipidémie, et/ou intolérance au glucose (16), la définition étant moins stricte que pour le syndrome métabolique.
Bien que ce syndrome soit proche et qu’il y ait un certain chevauchement avec la stéato-hépatite non alcoolique, l’hépatosidérose métabolique doit en être distinguée puisque seulement un tiers des patients présentant une stéato-hépatite non alcoolique a aussi une hépatosidérose métabolique, et qu’inversement seulement la moitié des patients présentant une hépatosidérose métabolique a une maladie stéatosique du foie (17). Les mécanismes en jeux sont mal définis, la régulation de l’hepcidine et de l’absorption du fer serait altérée (14) et des études in vitro suggèrent que la surcharge en fer des adipocytes influencerait de manière significative la sécrétion de différentes adipokines et donc l’insulino-résistance (2).
L’hyperferritinémie évolue autour de 800 µg/l et le coefficient de saturation de la transferrine est normal ou très légèrement augmenté. La surcharge en fer est habituellement modérée (concentration hépatique en fer mesurée par IRM < 100 µmol/g). Cette surcharge est cependant réelle puisque mobilisable par la réalisation de saignée permettant de soustraire 2,5±0,5 gramme de fer contre 0,8 ± 0,3 chez des sujets contrôles (18).
Hyperferritinémies secondaires sans surcharge
Deux grands mécanismes peuvent conduire à une augmentation, parfois majeure, de la ferritine sans qu’elle soit associée à une surcharge en fer : l’augmentation de sa synthèse, et l’augmentation de son relargage du milieu intracellulaire vers le plasma. Syndrome inflammatoire et maladie systémique : la ferritine est une protéine de la phase aiguë de l’inflammation, elle joue un rôle anti-inflammatoire par la séquestration du fer limitant ainsi le stress oxydant. Sa synthèse est fortement augmentée sous l’influence d’interleukines et du TNF-< (19). L’augmentation peut être marquée au cours d’un épisode infectieux aigu, comme de façon chronique lors de maladies de systèmes associées ou non à une augmentation de la CRP.
Cytolyse : la ferritine circulante représente à l’état physiologique une fraction faible de la ferritine contenue dans les cellules. Une souffrance cellulaire ou une nécrose de cellules riches en ferritine (hépatocyte, myocyte…) va entraîner un relargage de ferritine dans la circulation faussant sa représentativité du stock en fer intracellulaire. Un recyclage excessif de cellules riches en fer (Syndrome d’Activation macrophagique) peut aussi entraîner une forte hyperferritinémie sans surcharge en fer.
La consommation d’alcool peut être une source d’hyperferritinémie par des mécanismes indirects précédemment évoqués (stéato-hépatite, lyse hépatocytaire), mais aussi par des mécanismes directs. En effet l’alcool peut d’une part induire directement la synthèse de ferritine (20), et d’autre part inhiber l’hepcidine et ainsi être responsable d’une surcharge en fer secondaire modérée (21).
D’autres causes plus anecdotiques d’hyperferritinémie sans surcharge en fer associée peuvent être évoquées en deuxième ligne : syndrome paranéoplasique, hyperthyroïdies, maladie de Gaucher (organomégalie et atteinte osseuse), maladie de Still (fièvre, arthralgies et rash cutané).
Hyperferritinémies avec surcharges en fer secondaires
Maladie chronique du foie et cirrhose
À côté de l’hyperferritinémie induite par l’activité nécrotico-inflammatoire, la plupart des maladies chroniques du foie peuvent être associées à une surcharge en fer. Au stade précoce de la maladie du foie, cette surcharge mésenchymateuse minime est probablement liée au recyclage des cellules nécrotiques par les macrophages. Au stade de cirrhose, une surcharge en fer est trouvée chez 30 % des patients et peut être de localisation hépatocytaire ou mixte (22). Elle est proportionnelle à la sévérité de la cirrhose et peut parfois être massive, posant parfois la question de la causalité de la surcharge en fer dans des cirrhoses d’étiologies incertaines ou mixtes (23). Sa répartition est hétérogène d’un nodule à l’autre et la fibrose est indemne de fer, ce qui permet de la distinguer d’une surcharge en fer primitive. Il est probable que le mécanisme soit lié à l’insuffisance hépatocellulaire, la diminution de synthèse d’hepcidine entraînant une majoration de l’absorption du fer, et la baisse de synthèse de transferrine majore son taux de saturation et donc l’apparition de fer non liée à la transferrine.
Surcharge iatrogène
Plusieurs contextes de supplémentation en fer (parfois méconnue) peuvent conduire à d’authentiques surcharges en fer :
- Iatrogène, en général très facilement identifiable, que ce soit par le biais de prescription de supplémentation orale continuée sur de très longues périodes sans surveillance biologique, ou de pathologies inflammatoires chroniques au cours desquelles la prise en charge de l’anémie est particulièrement difficile et nécessite des supplémentations itératives (insuffisance rénale chronique, MICI) (24).
- Automédication, moins évidente à dépister, car parfois méconnue par les patients eux-mêmes, avec l’utilisation de compléments alimentaires parfois très riches en fer, ou d’utilisation abusive et chronique de « cures de vitamines ».
- La recherche d’une amélioration des performances sportives, cyclistes (25), musculation, avec prise chronique de fer par voie orale, de complément alimentaire enrichi en fer (Spiruline).
Maladies hématologiques
La transfusion d’un concentré érythrocytaire apporte 200 à 250 mg de fer, si cela ne pose aucun problème pour une transfusion anecdotique, cela peut conduire à une surcharge non négligeable, voire massive lors de transfusions importantes (leucémie, lymphome, greffe de moelle…) ou de transfusions chroniques (myélodysplasie, thalassémie…) (26, 27).
Bien que souvent négligée à la phase aiguë – en raison d’un contexte facilement compréhensible – cette surcharge est cependant responsable d’une surmortalité à long terme d’origine cardiaque et hépatique chez ces patients [un tiers des décès dans une série asiatique de myélodysplasie (28)]. Comme toutes les surcharges en fer, en l’absence d’excrétion physiologique du fer, elle peut persister pendant des décennies (29, 30).
À côté des surcharges induites par les transfusions, les anomalies chroniques de l’érythropoïèse sont susceptibles d’induire une surcharge en fer, même en l’absence de transfusion. L’hypoxie relative engendrée par l’anémie va inhiber l’hepcidine et majorer l’absorption du fer (31). De plus, la mise en évidence récente de l’érythroferonne (32) clarifie le lien direct qui existe entre érythropoïèse et métabolisme du fer (33). Secrétée par les érythroblastes, l’érythroferonne, par un mécanisme qui reste indéterminé, va profondément inhiber l’hepcidine permettant ainsi l’absorption importante de fer nécessaire à l’érythropoïèse. Une surcharge en fer peut donc se constituer indépendamment de toute transfusion dans toutes les situations chroniques d’érythropoïèse augmentée (érythropoïèse inefficace, hémolyse chronique).
La porphyrie cutanée tardive est liée à une diminution de l’activité d’une enzyme de synthèse de l’hème qui est inactivée de façon réversible par un processus fer-dépendant. Une hépatosidérose mixte peu marquée est trouvée dans 60 % à 70 % des cas. Le rôle des cofacteurs (alcool, hépatite C) dans l’expression de la pathologie est important. Bien qu’une mutation du gène HFE semble conférer une susceptibilité particulière, les mécanismes de la surcharge en fer restent très mal incompris. La martiale par saignée permet la régression des manifestations cutanées y compris chez les patients ayant un stock en fer normal.
Hyperferritinémie non hémochromatosique d’origine génétique
La ferritine est une nanocage constituée par l’assemblage de 24 sous-unités, chaînes lourdes et chaînes légères, dont les ratios respectifs peuvent varier. La synthèse de ses sous-unités se fait principalement sous l’influence du stock en fer intracellulaire. Des variants délétères peuvent compromettre cette régulation et conduire à une synthèse inappropriée de ferritine. Cela a été mis en évidence au niveau du gène codant pour la chaîne légère de la ferritine (le gène FTL), avec une atteinte qui peut se situer soit au niveau de son ARN messager soit de son premier exon. Dans le cadre d’un variant délétère au niveau de l’ARN, une cataracte précoce peut être associée (Syndrome hyperferritinémie-Cataracte), tandis qu’un variant délétère dans le premier exon induit une hyperferritinémie sans atteinte organique.
Il est primordial de noter que dans les deux cas il s’agit d’hyperferritinémie franche, mais sans aucune surcharge en fer et qu’il n’y a aucune autre conséquence clinique que le risque de cataracte. Il n’y a donc aucun traitement ou surveillance à mettre en place et il faut être parfaitement rassurant vis-à-vis du patient. Du fait de la transmission autosomique dominante, il est indispensable de dépister les apparentés pour leur éviter des explorations et/ou traitements superflus.
| Syndrome Métabolique | Cytolyse | Inflammation | Alcool | Maladie du foie | Iatrogène | Hématologie | FTL |
| Ferritine | ↑ | ↑-↑↑ | ↑-↑↑ | ↑ | ↑ | ↑ | ↑-↑↑ | ↑↑ |
| Saturation | N (↑) | N -↓ | ↓↓ | N (↑) | N-↑↑ | N -↓ | ↑-↑↑ | N -↓ |
| CHF | N (↑) | N | N | N-↑↑ | N-↑↑ | ↑-↑↑ | ↑-↑↑ | N |
| CHF | Clinique | ASAT/
ALAT | CRP | Clinique | Clinique | Clinique | NFS,
réticulocytes… | Cataracte |
Récapitulatif des principales causes d’hyperferritinémies hors hémochromatose
Démarche diagnostique
Examens de première ligne
Examen clinique
C’est une étape fondamentale, trop souvent négligée, pour l’orientation correcte de la démarche diagnostique et pour éviter des explorations inappropriées et/ou inutiles.
L’interrogatoire cherche une cause de surcharge en fer secondaire :
L’interrogatoire recherche des éléments en faveur d’un syndrome génétique :
- Histoire familiale de surcharge en fer, en essayant d’obtenir des informations sur les facteurs confondants éventuels présents chez les apparentés
- Histoire familiale de cataracte précoce (syndrome hyperferritinémie cataracte)
L’examen clinique recueille le tour de taille (à défaut le poids et la taille) et la tension artérielle à la recherche d’un syndrome métabolique.
Examens biologiques
Confirmer les perturbations sur un deuxième bilan
Il existe une variation circadienne (maximum le matin) de la saturation de la transferrine, ainsi que d’un jour à l’autre. Par ailleurs, il est nécessaire de vérifier le taux sérique de transferrine pour juger de la fiabilité du coefficient de saturation.
En raison de ces nombreux facteurs confondants, il faut réaliser un contrôle à distance du bilan martial (ferritine et coefficient de saturation de la transferrine), dans un délai adapté à la situation clinique (sevrage ?), pour juger de la stabilité des anomalies. D’autre part, en l’absence de standardisation du dosage de la ferritine il faut toujours interpréter ses valeurs en fonction des seuils de la technique du laboratoire.
Prendre en compte les facteurs confondants
De façon systématique lors d’une hyperferritinémie confirmée, le bilan biologique doit être complété pour prendre en compte les causes de surcharges en fer acquises et les hyperferritinémies secondaires sans lien avec le métabolisme du fer :
- Bilan lipidique, et glycémie à jeun : recherche d’un syndrome métabolique.
- Protéine C réactive : éliminer un syndrome inflammatoire, ainsi que le bilan hépatique et les CPK à la recherche d’une lyse cellulaire.
- Bilan hépatique (ASAT, ALAT, GGT, PAL) à la recherche d’une maladie chronique du
- Numération formule sanguine, haptoglobine, réticulocytes : recherche d’une maladie hématologique, anémie chronique ou hémolyse (parfois compensée).
Examens de deuxième ligne
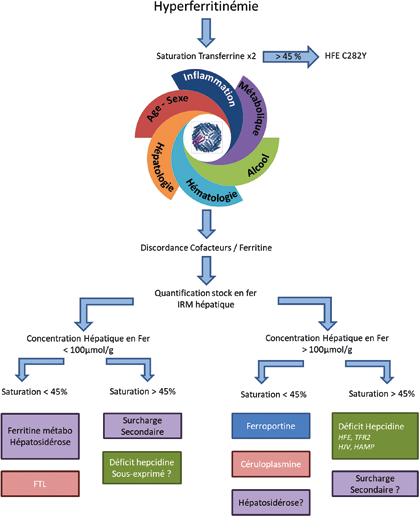
En l’absence de cause évidente de surcharge en fer secondaire, et devant la confirmation d’une hyperferritinémie non simplement expliquée par les facteurs confondant, la suite de la démarche diagnostique est guidée par le niveau de saturation de la transferrine (qui doit être contrôlé sur un second prélèvement).
Saturation de la transferrine élevée
Rechercher une hémochromatose HFE
Une saturation de la transferrine élevée doit faire rechercher une hémochromatose HFE et donc le variant C282Y du gène HFE. L’homozygotie pose le diagnostic d’hémochromatose HFE classique. S’agissant d’une pathologie récessive, une simple hétérozygotie ne permet pas de poser un diagnostic. La recherche des variants non délétères H63D, et S65C, qui n’ont pas de conséquence pathologique n’est pas pertinente et n’est pas prise en charge par la sécurité sociale.
Quantification du stock en fer
En l’absence d’homozygotie HFE C282Y il faut avant d’aller plus loin objectiver la sévérité de la surcharge en fer. La méthode de référence est l’IRM hépatique qui permet une quantification simple et fiable de la concentration hépatique en fer (34).
En cas de discordance et/ou de doute sur la fiabilité de l’IRM, le stock en fer de l’organisme peut être estimé par le biais des saignées. Considérant qu’un litre de sang soustrait contient 0,5 g de fer, on peut a posteriori calculer le nombre de grammes de fer qu’il a fallu soustraire pour obtenir la désaturation (définie par une ferritine <50-100 µg/L). Cette méthode n’est fiable que si le délai entre deux saignées est inférieur à 15 jours, au-delà, l’absorption du fer dans l’intervalle fausse le résultat. Chez un sujet normal, on peut approximativement soustraire 1 à 2 g de fer.
La biopsie hépatique n’est pertinente dans cette optique que dans de rares cas de contre-indication à l’IRM et aux saignées. Elle peut toutefois être indiquée à des fins étiologiques en cas de maladie hépatique associée/sous-jacente. Elle permet alors une évaluation semi-quantitative du fer selon des classifications adaptées (23, 35), ces dernières sont cependant de moins en moins maîtrisées par les anatomopathologistes.
En l’absence de surcharge significative (autour de 100-120 µmol/g, 3-4 g de fer soustrait) il n’est (dans l’immense majorité des cas) pas justifié de pousser plus loin les investigations génétiques en raison de leur coût et de la faible probabilité d’une positivité. Il est en effet probable qu’il s’agisse de perturbations biologiques secondaires.
Rechercher une cause génétique rare de surcharge en fer
En présence d’une surcharge significative, une hémochromatose non HFE C28Y doit être évoquée. La saturation de la transferrine élevée oriente vers un mécanisme de déficit en hepcidine et classiquement deux groupes de gènes étaient distingués en fonction de l’âge du patient :
Si < 30 ans, une hémochromatose juvénile est la plus probable, les gènes impliqués sont HJV (hémojuvéline) et HAMP (hepcidine).
Chez un patient plus âgé, les gènes pouvant être impliqués sont HFE (variants délétères rares), TFR2 (Récepteur 2 de la transferrine), et SLC40A1 (Ferroportine, dans sa forme plus rare dite B).
Cependant cette distinction est remise en cause, d’une part en raison de présentations plus tardives ou au contraire plus précoces qu’attendues, d’autre part, car l’avènement du séquençage haut débit fait que les gènes ne sont plus étudiés un par un, mais par « panel » de gènes ciblé sur un syndrome de telle sorte que devant une surcharge en fer avec saturation élevée ces cinq gènes seraient séquencés simultanément par le laboratoire génétique moléculaire.
En cas de doute sur la nécessité de poursuivre ou non les investigations, le dossier doit être discuté auprès d’un des centres du réseau du centre national de références des hémochromatoses. (liste sur : https://centre-reference-fer-rennes.org/les-centres-de- competences)
Saturation normale ou basse
Les orientations diagnostiques sont ici aussi fonction de la présence ou non d’une surcharge en fer. La problématique réside dans la décision de poursuivre ou non les explorations. En effet, il n’existe pas de critère universel de niveau de ferritine justifiant d’évaluer le stock en fer, celle-ci doit donc être jugée à l’aune du contexte clinique et des explorations de 1re ligne. Selon toute vraisemblance, un seuil inférieur à deux fois la limite supérieure de la technique du laboratoire ne justifie pas de poursuivre les explorations.

Arbre diagnostique
Concentration hépatique en fer élevée
Évoquer une hépatosidérose métabolique
Elle est de très loin la situation la plus fréquente, le syndrome métabolique associé peut être plus ou moins prononcé et parfois même être incomplet. La surcharge en fer est habituellement modérée (CHF <100 µmol/g ou grammes de fer soustraits ± 2,5 g), de même que la ferritine (800 ± 200 µg/L), mais toutes deux peuvent parfois être plus importantes. Il s’agit d’un syndrome clinique dont le diagnostic se fait par élimination.
Recherche une cause génétique rare de surcharge en fer à saturation normale
La maladie de la Ferroportine doit être évoquée. Cette forme rare d’hémochromatose est liée à l’atteinte du transporteur (le seul connu) permettant l’export du fer par les cellules. Le phénotype classique est superposable à celui d’une hépatosidérose métabolique, cependant la surcharge en fer est en général plus importante et peut parfois être massive. La distinction Ferroportine / hépatosidérose métabolique est difficile à faire et il n’existe pas à ce jour de recommandation permettant de statuer sur le niveau de ferritine ou de surcharge en fer à partir duquel le test génétique doit être demandé. S’agissant d’une maladie autosomique dominante, une histoire familiale de surcharge en fer est un fort argument en faveur de la réalisation du test.
Une autre forme encore plus rare de cause génétique au profil atypique, et en général évocateur peut être envisagée : l’acéruloplasminémie qui associe à des degrés variables une anémie microcytaire, une atteinte neurologique (ataxie, dysarthrie, mouvements anormaux), un diabète, une rétinopathie. Un simple dosage de la céruloplasmine oriente le diagnostic.
En cas de doute sur la nécessité de réaliser un test génétique, un avis complémentaire peut être pris auprès d’un des centres du réseau du centre national de références des hémochromatoses.
Concentration hépatique en fer normale
Deux causes d’hyperferritinémie sont à évoquer : l’hyperferritinémie métabolique et une anomalie du gène FTL. L’hyperferritinémie métabolique est de très loin la situation la plus fréquente. Le syndrome métabolique associé est plus ou moins prononcé, et l’hyperferritinémie peut précéder l’apparition du syndrome métabolique (cf. supra).
Le seuil de ferritine à partir duquel une analyse du gène FTL doit être demandée n’est pas établi, une fois encore la ferritine doit être interprétée à l’aune du contexte clinique et des explorations de 1re ligne. C’est en particulier la discordance entre l’augmentation de la ferritine avec la concentration hépatique en fer qui est évocatrice. Ainsi une ferritine à 800 µg/L avec une CHF à 24 µmol/g sera plus évocatrice qu’une ferritine à 1 000 µg/L avec une CHF à 70 µmol/g.
En cas de doute sur la nécessité de réaliser un test génétique, un avis complémentaire peut être pris auprès d’un des centres du réseau du centre national de références des hémochromatoses.
Prise en Charge Dépistage familial
Lors de la mise en évidence d’une pathologie génétique, il est indispensable de considérer le dépistage familial. Les apparentés au premier degré doivent être informés par le patient de la pathologie concernée et des modalités de dépistage. Cela permet un diagnostic plus précoce qui est associé à un moindre retentissement organique grâce à la prise en charge. Dans d’autres déplétion situations, cela permet d’être rassurant et d’éviter aux apparentés des explorations et suivis inutiles (par exemple FTL).
Traitement
Quelles modalités ?
Le traitement vise dans un premier temps à normaliser le stock en fer, puis dans certaines situations à prévenir la reconstitution de la surcharge en fer.
Saignées
Les saignées sont le traitement de choix de la surcharge en fer. La poussée d’érythropoïèse secondaire à la perte sanguine va consommer une quantité significative de fer permettant par sa répétition de normaliser le stock. Afin d’obtenir une baisse significative, il faut que les saignées soient suffisamment rapprochées, car l’absorption du fer est elle aussi stimulée après une saignée.
C’est un traitement simple et bien toléré, qui peut être réalisé au domicile du patient par une infirmière une fois que les 5 premières saignées en structures de soins se sont déroulées de façon satisfaisante. Le volume soustrait est habituellement de 7 ml/kg sans dépasser 550 ml par saignée. L’utilisation de ces saignées pour le don du sang a longtemps été complexe. Les dispositions réglementaires du don-saignée, ont récemment évolué et permettent maintenant leur réalisation dans tous les sites fixes de collecte de sang. Le suivi clinique et biologique se fait sous la responsabilité du prescripteur et la compatibilité avec les critères de sélection des donneurs est vérifiée lors du don-saignée.
Le traitement par saignée est contre indiqué, ou à discuter, avec le spécialiste référent en cas de pathologie cardio-vasculaire ou hématologique associée.
En dehors de la surveillance clinique habituelle il est recommandé de contrôler la ferritine mensuellement jusqu’à sa normalisation (300 µg/L chez l’homme, 200 µg/L chez la femme) puis toutes les deux saignées une fois ces valeurs atteintes. La surveillance biologique se fait sur le même prélèvement que la saignée. Au cours du traitement de maintenance, la surveillance se fait toutes les deux saignées.
La périodicité de la surveillance de la numération formule sanguine peut suivre celle de la ferritine.
Dans les surcharges d’origine génétique, l’objectif est habituellement de viser une ferritine à 50 µg/L par analogie à l’hémochromatose HFE, mais cela doit être adapté à la tolérance et la situation clinique. Ainsi pour les maladies de la Ferroportine à saturation de la transferrine normale, le seuil est habituellement plus élevé.
Chélateurs
Trois chélateurs du fer sont actuellement disponibles: la déféroxamine, le déférasirox et la défériprone. Initialement développés pour les maladies hématologiques, car les saignées sont alors souvent contre-indiquées, ils ont aussi une place pour les patients ayant d’autres causes de surcharge et chez qui les saignées ne sont pas réalisables pour des raisons techniques (abord veineux), ou médicales. Dans certaines surcharges génétiques massives (hémochromatose juvénile) il peut être intéressant de les associer aux saignées pour raccourcir la durée de désaturation.
La déféroxamine est le traitement le plus ancien et pour lequel le plus de données sont disponibles. Il est le seul à avoir une autorisation de mise sur le marché dans le cadre d’une hémochromatose. Il pose cependant un problème d’observance, car doit être administré par voie sous-cutanée en continu ce qui le rend peu pratique. Il existe une toxicité potentielle au niveau cardiaque, rénal, auditif et ophtalmologique qui nécessite une surveillance.
Le déférasirox présente l’avantage d’être administré par voie orale en une prise quotidienne. Il dispose d’une autorisation de mise sur le marché pour les surcharges hématologiques, mais pas pour l’hémochromatose. Un essai de phase I/II dans l’hémochromatose HFE a suggéré sa bonne tolérance et son efficacité (36). Les principaux effets secondaires sont cutanés, rénaux et gastro-intestinaux.
Le défériprone est aussi administré par voie orale, en deux prises, et avec des autorisations de mise sur le marché similaires à celle du déférasirox. Il n’y a pas de donnée sur son utilisation dans l’hémochromatose, mais il présente la particularité de franchir la barrière hémato encéphalique, ce qui est intéressant dans les maladies avec atteinte neurologique (acéruloplasminémie). Ses principaux effets secondaires sont un risque d’agranulocytose, des troubles gastro-intestinaux, et des douleurs articulaires.
Quelle place pour le régime pauvre en fer ?
Bien que cela soit souvent une préoccupation importante des patients, le régime ne semble pas avoir une importance significative dans la prise en charge des surcharges en fer.
Il faut rappeler que seulement une fraction minime (± 10 %) du fer présent dans l’alimentation est finalement absorbée au niveau du tube digestif, de telle sorte que s’il existe un déficit en hepcidine induisant une hyper absorption une surcharge se constituera même avec un apport en fer limité.
Il faut toutefois appeler les patients à la vigilance vis-à-vis d’éventuels compléments alimentaires qui peuvent contenir du fer, ou la prise régulière de vitamine C qui favorise fortement son absorption.
Il n’y a pas de données de qualité disponible, cependant certains patients particulièrement investis dans leur alimentation peuvent arriver à diminuer légèrement leur besoin en saignée dans le cadre d’une hémochromatose HFE. Cela doit toutefois relever d’un choix personnel du patient plus que d’une recommandation médicale.
Quelles indications ?
Surcharges en fer d’origine génétique
Bien qu’il n’y ait pas de preuve scientifique significative, et par analogie à l’hémochromatose HFE dont le mécanisme physiopathologique et le phénotype sont identiques, il est nécessaire de traiter efficacement les surcharges d’origines génétiques associées à un déficit en hepcidine (37). En effet, la saturation de la transferrine haute signe une exposition aux formes toxiques de fer qui vont induire les lésions organiques (38). Les saignées sont la méthode de choix.
Dans les surcharges génétiques sans déficit en hepcidine (Ferroportine), il n’y a pas de donnée permettant d’évaluer la pertinence d’un traitement déplétif ou l’évolution à long terme de ces patients (39). La saturation de la transferrine normale suggère l’absence d’exposition à des formes toxiques de fer, et les séries de patients décrites ne font pas état d’un retentissement organique clair (40). Par principe de précaution un traitement par saignée est proposé pour tenter de normaliser le stock en fer. Les saignées ne sont pas toujours bien tolérées et doivent donc être proposées avec des volumes et une périodicité plus faible que dans l’hémochromatose HFE. En l’absence de preuve formelle du bénéfice, la balance bénéfice risque d’un traitement médicamenteux (chélateur) doit être soigneusement évaluée.
Traitement de l’hépatosidérose métabolique
Un certain nombre d’études cliniques – de méthodologie et/ou de taille souvent limitée – ont suggéré un effet bénéfique de la réalisation de saignée sur l’insulino-résistance (41-44). Ceci allant dans le sens de l’effet délétère du fer sur les métabolismes glucidiques et lipidiques in vitro. Il a donc longtemps été préconisé la mise en place d’un traitement déplétif chez les patients ayant une hépatosidérose métabolique.
Cependant deux travaux récents ayant inclus un nombre significatif de patients, et avec une méthodologie de qualité, ont démontré l’absence de bénéfice d’un traitement par saignée dans le cadre de l’hépatosidérose métabolique (45) ou d’une stéato-hépatite non alcoolique (46). Bien que certains éléments de ces travaux puissent être discutés (durée de suivi, population étudiée, modalité de traitement) il ne semble pas pertinent de proposer un traitement déplétif dans ce contexte.
Traitement des surcharges secondaires
Les surcharges en fer secondaire aux maladies hématologiques sont associées à une surmortalité. Il est donc logique et recommandé de mettre en place un traitement déplétif en fonction de l’espérance de vie (47, 48). Dans ce contexte les saignées sont rarement réalisables, et les traitements chélateurs sont alors l’option de choix. L’objectif est en général moins strict que dans l’hémochromatose avec une ferritine cible <500 µg/L.
La prise en charge d’une porphyrie cutanée tardive repose sur l’éviction des facteurs favorisants et la mise en place d’un traitement déplétif qui sera poursuivi au long cours.
Il n’y a pas de données suffisantes pour établir des recommandations concernant les autres causes de surcharges secondaires.
La prise en charge se discute en fonction des possibilités thérapeutiques et du contexte. La présence d’une saturation de la transferrine augmentée est en faveur de l’initiation d’un traitement pour limiter l’exposition aux formes toxiques de fer.
Cependant de nombreux contextes ne sont pas compatibles avec un traitement (cirrhose grave, insuffisance rénale chronique dialysée).
Traitement des hyperferritinémies sans surcharge
Il n’y a en l’absence de surcharge en fer aucune indication à mettre en place un traitement déplétif d’une hyperferritinémie.
Surveillance
Une fois le diagnostic établi se pose la question de la surveillance de l’hyperferritinémie. Il est capital de distinguer deux situations :
- Surcharge en fer (génétique ou secondaire) indiquant un traitement :
Il est alors nécessaire de surveiller l’évolution du stock en fer de l’organisme pour adapter le traitement, et la ferritine devra donc être contrôlée régulièrement. Le rythme sera à adapter au type et à la périodicité du traitement ou à l’évolution spontanée de la ferritine chez les patients qui ne sont pas traités.
- Hyperferritinémie sans surcharge significative
Dans cette situation, une fois le diagnostic posé, il n’y a pas intérêt à effectuer une surveillance de la ferritine. Il n’y a en effet aucune donnée scientifique permettant de donner une valeur informative à l’augmentation ou la baisse de la ferritine au cours du suivi. Il n’y a en particulier pas d’élément indiquant si une amélioration du syndrome métabolique (par exemple une perte de poids) est sensée entraîner une baisse de la ferritine. N’ayant aucune conséquence pratique, il est donc inutile de répéter un dosage qui risque de détourner l’attention du patient de la problématique réelle.
Références
- Diouf I, Charles MA, Ducimetiere P et al. Evolution of obesity prevalence in France: an age-period-cohort Epidemiology 2010;21:360-5.
- Gabrielsen JS, Gao Y, Simcox JA et al. Adipocyte iron regulates adiponectin and insulin J Clin Invest 2012;122:3529-40.
- Seldin MM, Peterson JM, Byerly MS et al. Myonectin (CTRP15), a novel myokine that links skeletal muscle to systemic lipid homeostasis. J Biol Chem 2012;287:11968-80.
- Simcox JA, McClain DA. Iron and diabetes risk. Cell Metab 2013;17:329-41.
- Park SK, Ryoo JH, Kim MG et Association of serum ferritin and the development of metabolic syndrome in middle-aged Korean men: a 5-year follow-up study. Diabetes Care 2012;35:2521-6.
- Podmore C, Meidtner K, Schulze MB et Association of Multiple Biomarkers of Iron Metabolism and Type 2 Diabetes: The EPIC-InterAct Study. Diabetes Care 2016;39:572-81.
- Ellervik C, Marott JL, Tybjaerg-Hansen A et Total and cause-specific mortality by moderately and markedly increased ferritin concentrations: general population study and metaanalysis. Clin Chem 2014;60:1419-28.
- Hagstrom H, Nasr P, Bottai M et al. Elevated serum ferritin is associated with increased mortality in non-alcoholic fatty liver disease after 16 years of follow-up. Liver Int 2016;36:1688-1695.
- Ford ES et Cogswell ME. Diabetes and serum ferritin concentration among U.S. adults. Diabetes Care 1999;22:1978-83.
- Fernandez-Real JM, Ricart-Engel W, Arroyo E et al. Serum ferritin as a component of the insulin resistance Diabetes Care 1998;21:62-8.
- Jehn M, Clark JM et Guallar E. Serum ferritin and risk of the metabolic syndrome in U.S. adults. Diabetes Care 2004;27:2422-8.
- Kowdley KV, Belt P, Wilson LA et al. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2012;55:77-85.
- Valenti L, Fracanzani AL, Bugianesi E et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2010;138:905-12.
- Rametta R, Dongiovanni P, Pelusi S et al. Hepcidin resistance in dysmetabolic iron overload. Liver Int 2016;36:1540-8.
- Nelson JE, Wilson L, Brunt EM et al. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011;53:448-57.
- Moirand R, Mortaji AM, Loreal O et al. A new syndrome of liver iron overload with normal transferrin Lancet 1997;349:95-7.
- Mendler MH, Turlin B, Moirand R et al. Insulin resistance-associated hepatic iron Gastroenterology 1999;117:1155-63.
- Jezequel C, Laine F, Laviolle B et al. Both hepatic and body iron stores are increased in dysmetabolic iron overload syndrome. A case-control PLoS One 2015;10:e0128530.
- Torti FM et Torti Regulation of ferritin genes and protein. Blood 2002;99:3505-16.
- Moirand R, Kerdavid F, Loreal O et al. Regulation of ferritin expression by alcohol in a human hepatoblastoma cell line and in rat hepatocyte cultures. J Hepatol 1995;23:431-9.
- Harrison-Findik DD, Klein E, Crist C et al. Iron-mediated regulation of liver hepcidin expression in rats and mice is abolished by alcohol. Hepatology 2007;46:1979-85.
- Ludwig J, Hashimoto E, Porayko MK et al. Hemosiderosis in cirrhosis: a study of 447 native Gastroenterology 1997;112:882-8.
- Deugnier Y ett Turlin B. Pathology of hepatic iron overload. World J Gastroenterol 2007;13:4755-60.
- Rostoker G, Griuncelli M, Loridon C et al. Hemodialysis-associated hemosiderosis in the era of erythropoiesis-stimulating agents: a MRI Am J Med 2012;125:991-999 e1.
- Deugnier Y, Loreal O, Carre F et al. Increased body iron stores in elite road cyclists. Med Sci Sports Exerc 2002;34:876-80.
- Taher AT et Saliba Iron overload in thalassemia: different organs at different rates. Hematology Am Soc Hematol Educ Program 2017;2017:265-271.
- Shenoy N, Vallumsetla N, Rachmilewitz E et al. Impact of iron overload and potential benefit from iron chelation in low-risk myelodysplastic syndrome. Blood 2014;124:873-81.
- Takatoku M, Uchiyama T, Okamoto S et al. Retrospective nationwide survey of Japanese patients with transfusion- dependent MDS and aplastic anemia highlights the negative impact of iron overload on morbidity/mortality. Eur J Haematol 2007;78:487-94.
- Nottage K, Gurney JG, Smeltzer M et al. Trends in transfusion burden among long-term survivors of childhood hematological malignancies. Leuk Lymphoma 2013;54:1719-23.
- Sirvent A, Auquier P, Oudin C et al. Prevalence and risk factors of iron overload after hematopoietic stem cell transplantation for childhood acute leukemia: a LEA study. Bone Marrow Transplant 2017;52:80-87.
- Mastrogiannaki M, Matak P, Delga S et al. Deletion of HIF-2alpha in the enterocytes decreases the severity of tissue iron loading in hepcidin knockout mice. Blood 2012;119:587-90.
- Kautz L, Jung G, Valore EV et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet 2014;46:678-84.
- Kautz L, Jung G, Nemeth E et al. Erythroferrone contributes to recovery from anemia of inflammation. Blood 2014;124:2569-74.
- d’Assignies G, Paisant A, Bardou-Jacquet E et al. Non-invasive measurement of liver iron concentration using 3-Tesla magnetic resonance imaging: validation against biopsy. Eur Radiol 2018;28:2022-2030.
- Scheuer PJ, Williams R et Muir Hepatic pathology in relatives of patients with haemochromatosis. J Pathol Bacteriol 1962;84:53-64.
- Phatak P, Brissot P, Wurster M et al. A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology 2010;52:1671-779.
- EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol 2010;53:3-22.
- Brissot P, Ropert M, Le Lan C et al. Non-transferrin bound iron: a key role in iron overload and iron toxicity. Biochim Biophys Acta 2012;1820:403-10.
- Mayr R, Janecke AR, Schranz M et Ferroportin disease: a systematic meta-analysis of clinical and molecular findings. J Hepatol 2010;53:941-9.
- Le Lan C, Mosser A, Ropert M et al. Sex and acquired cofactors determine phenotypes of ferroportin Gastroenterology 2011;140:1199-1207 e1-2.
- Valenti L, Fracanzani AL, Dongiovanni P et al. Iron depletion by phlebotomy improves insulin resistance in patients with nonalcoholic fatty liver disease and hyperferritinemia: evidence from a case-control Am J Gastroenterol 2007;102:1251-8.
- Fernandez-Real JM, Penarroja G, Castro A et Blood letting in high-ferritin type 2 diabetes: effects on insulin sensitivity and beta-cell function. Diabetes 2002;51:1000-4.
- Beaton MD, Chakrabarti S, Levstik M et al. Phase II clinical trial of phlebotomy for non-alcoholic fatty liver Aliment Pharmacol Ther 2013;37:720-9.
- Facchini FS, Hua NW et Stoohs RA. Effect of iron depletion in carbohydrate-intolerant patients with clinical evidence of nonalcoholic fatty liver disease. Gastroenterology 2002;122:931-9.
- Laine F, Ruivard M, Loustaud-Ratti V et al. Metabolic and hepatic effects of bloodletting in dysmetabolic iron overload syndrome: A randomized controlled study in 274 patients. Hepatology 2017;65:465-474.
- Adams LA, Crawford DH, Stuart K et al. The impact of phlebotomy in nonalcoholic fatty liver disease: A prospective,randomized, controlled trial. Hepatology 2015;61:1555-64.
- Maggio A. Light and shadows in the iron chelation treatment of haematological diseases. Br J Haematol 2007;138:407-21.
- Angelucci E, Barosi G, Camaschella C et al. Italian Society of Hematology practice guidelines for the management of iron overload in thalassemia major and related disorders. Haematologica 2008;93:741-52.