LIENS D’INTÉRÊTS
Board/consultant : Amgen, AstraZeneca, Baxter, Bristol-Myers Squibb, Fresenius Kabi, Incyte Biosciences, Merck, MSD, Mylan, Novartis, Nutricia, Pierre Fabre, Roche, Sanofi, Servier
Financement recherche : Roche
Essais cliniques : AstraZeneca, Bristol-Myers Squibb, OSE Immunotherapeutics
MOTS-CLÉS
biomarqueurs, médecine personnalisée, thérapies ciblées
ABRÉVIATIONS
ADNtc : ADN tumoral circulant, AMM : autorisation de mise sur marché CCA : cholangiocarcinome, CCR : cancer colorectal, CIMP : CpG Island Methylation Phenotype, CIN : Chromosomal INstability, CMS : Consensus Molecular Subtypes, CTLA4 : cytotoxic T-lymphocyte associated protein 4, CVB : cancers des voies biliaires EBV : virus Epstein-Barr, EGFR : récepteur du facteur de croissance épidermique, ESCAT : ESMO Scale for Clinical Actionability of molecular Targets ESMO : European Society of Medical Oncology, FGFR : récepteur au facteur de croissance des fibroblastes, GIST : tumeurs stromales gastro-intestinales HR : Hazard ratio, HRD : réparation de l’ADN par recombinaison homologue IDH1 : isocitrate déshydrogenase-1, IHC : immunohistochimie IMK : inhibiteur multikinase ISH : hybridation in situ, JOG : jonction oeso-gastrique mAc : anticorps monoclonal, MAPK : mitogen activated protein kinase, MLH1 : mutL homologue 1 MMR : MisMatch Repair MSH2 : mutS homologue 2 MSH6 : mutS homologue 6 MSI : microsatellites instables, NIH : National Institute of Health NGS : Next Generation Sequencing PARP : poly(ADP-ribose) polymérase, PCR : réaction d’amplification en chaîne par polymérase, PD1 : programmed death-1, PD-L1 : programmed death-1 ligand, PMS2 : postmeiotic segregation increased 2, SG : survie globale, SSM : survie sans maladie SSP : survie sans progression, T-DM1 : trastuzumab-emtansine, TNCD : Thesaurus National de Cancérologie Digestive TRK : récepteur kinase de la tropomyosine
Introduction
Jusqu’aux années 1990, la chimiothérapie cytotoxique était l’unique outil thérapeutique médical des cancers digestifs. Ces agents thérapeutiques affectent préférentiellement les cellules à division rapide, y compris les cellules cancéreuses, mais également les tissus normaux hautement prolifératifs (notamment les muqueuses digestives et la moelle osseuse), ce qui entraîne des toxicités fréquentes et limitantes. Une meilleure compréhension de la biologie moléculaire des cellules cancéreuses a conduit à la révolution thérapeutique des thérapies dites « ciblées », c’est-à- dire des anticorps monoclonaux (mAc) ou de petites molécules inhibitrices multikinases (IMK), dirigées contre des protéines spécifiquement soit surexprimées, soit mutées dans les cellules cancéreuses (1).
Les thérapies ciblées ont apporté un bénéfice clinique dans de nombreux types de cancers, notamment digestifs (1, 2). Cependant, leurs effets ont parfois été décevants, en raison de mécanismes de résistance intrinsèque ou acquise, d’une activité limitée à certains contextes tumoraux ou sous-groupes moléculaires, ou d’un profil de toxicités moins favorable qu’attendu. Ces échecs ont mis en évidence l’importance d’une sélection
rigoureuse des patients et ont révélé la complexité de la biologie des cancers digestifs à la fois au niveau inter-tumoral et également au sein d’un type de tumeur donné (1, 2).
L’identification de biomarqueurs prédictifs de la réponse à ces thérapies est donc devenue un enjeu majeur et l’ère de la médecine personnalisée s’est ouverte. D’un traitement standard pour tous les patients dans une indication donnée, des protocoles se sont développés spécifiquement pour certains sous-types de tumeurs, sur la base de l’identification de biomarqueurs prédictifs.
Selon le National Institute of Health (NIH), le terme de biomarqueur désigne « une caractéristique qui est mesurée et évaluée objectivement comme
indicateur d’un processus biologique normal ou pathologique, ou traduisant la réponse pharmacologique à une intervention thérapeutique » (3). Définis comme tels, les biomarqueurs peuvent s’appliquer à de nombreux domaines cliniques et peuvent être de natures très variées : cliniques, moléculaires (sanguins ou tumoraux) ou d’imagerie.
En cancérologie, trois catégories de biomarqueurs sont distinguées (4) :
- les marqueurs diagnostiques, correspondant aux tests permettant d’aider au diagnostic ou à l’identification de sous-classes d’une maladie particulière ;
- les marqueurs pronostiques, qui sont associés à une évolution de la maladie plus ou moins favorable en termes de survie sans maladie (SSM, après chirurgie) ou sans progression (SSP, stades avancés) et/ou globale (SG), indépendamment des traitements administrés (« histoire naturelle ») ;
- les marqueurs prédictifs, qui prédisent l’activité d’un traitement spécifique et ont vocation à être utilisés comme outils d’aide à la décision thérapeutique ; lorsqu’ils permettent de sélectionner les patients pour un traitement, ils sont qualifiés de « tests compagnons » (5).
Nous nous concentrerons ici plus particulièrement sur les biomarqueurs moléculaires tumoraux prédictifs, influençant la réponse aux traitements (ciblés ou non). L’analyse des biomarqueurs est effectuée sur un fragment de tumeur, fixé et inclus en paraffine le plus souvent ou congelé (biopsie ou pièce de résection chirurgicale, tumeur primitive ou métastase). Ils peuvent être étudiés par des techniques de biologie moléculaire (réaction d’amplification en chaîne par polymérase [PCR], ex. : mutations KRAS), d’immunohistochimie (IHC) et/ou d’hybridation in situ (ISH, ex. : surexpression/amplification HER2). Ces dernières années, l’avènement des techniques de séquençage à haut débit (Next Generation Sequencing, NGS) a également élargi la dimension de la recherche de ces anomalies, passant de leur recherche individuelle à l’étude de panels pouvant atteindre plusieurs centaines de gènes. Publiée en 2018, la classification ESCAT (ESMO Scale for Clinical Actionability of molecular Targets) proposée par l’European Society of Medical Oncology (ESMO) permet de classer ces altérations moléculaires selon leur « actionnabilité », c’est-à-dire la démonstration d’un bénéfice clinique obtenu par leur ciblage thérapeutique, allant de I (prêt pour la pratique de routine) à IV (rationnel pré-clinique) en fonction du niveau de preuve (Tableau 1) (6).
Dans cette mise au point, nous présenterons les principales anomalies moléculaires influençant la décision en oncologie digestive (ESCAT I/II (7) et/ou référencées dans le Thésaurus National de Cancérologie Digestive [TNCD]), les indications de la caractérisation moléculaire en oncologie digestive, et les modalités pratiques de réalisation d’une caractérisation moléculaire.
| Classe ESCAT | Valeur clinique | Implication clinique |
|---|
| Prêt pour l’utilisation en routine | I : Couple altération moléculaire et traitement ciblé associé à un bénéfice dans des essais cliniques | Le traitement ciblé administré aux patients présentant l’altération moléculaire a montré une amélioration des résultats cliniquesdans les essais cliniques prospectifs (randomisés ou non, études basket/multi-tumeurs, avec données de survie) | L’accès au traitement doit être considéré comme la référence thérapeutique |
| Expérimental | II : Couple altération moléculaire et traitement ciblé associé à une activité anti-tumorale, mais l’amplitude du bénéfice est inconnue | Le traitement ciblé administré à des patients définis sur le plan moléculaire est susceptible d’améliorer les résultats cliniques dans un type de tumeur (étude rétrospective, ou prospective
sans donnée de survie), mais des données supplémentaires sont nécessaires | Traitement à évaluer prospectivement (registre ou essai clinique) |
| Cible hypothétique | III : Couple altération moléculaire et traitement ciblé présumé améliorer les résultats cliniques sur la base d’essais cliniques dans d’autres types de tumeurs avec la même altération | Le traitement ciblé a précédemment été démontré bénéfique pour un sous-ensemble moléculaire défini dans un autre type de tumeur (ou avec une mutation différente dans le même gène), l’efficacité est donc
présumée mais non prouvée | Essai clinique à discuter avec les patients |
| IV : Actionnabilité pré-clinique démontrée | L’actionnabilité est présumée sur la base d’études précliniques, aucune donnée clinique concluante disponible | Le traitement ne doit être envisagé que dans le cadre d’essais cliniques précoces ; le manque de données cliniques doit être souligné auprès des patients |
| Association de traitements | V : Couple altération moléculaire et traitement ciblé associé à une réponse objective, mais sans bénéfice cliniquement significatif | Le médicament est actif mais ne prolonge pas la SSP ou la SG, probablement en partie en raison de
mécanismes d’adaptation | Des essais cliniques évaluant les stratégies d’association de traitements pourraient être envisagés |
Tableau 1 : Classification ESCAT de l’European Society of Medical Oncology [d’après Mateo et al. (6)]
Cancer colorectal (CCR)
Instabilité des microsatellites
Les progrès de la biologie moléculaire ont permis d’individualiser trois principaux mécanismes de cancérogenèse colorectale : (1) l’instabilité chromosomique (CIN : Chromosomal INstability) impliquée dans environ 80 % des CCR ; (2) l’instabilité microsatellitaire (MSI : microsatellites instables) impliquée dans 12 % à 15 % des CCR ; (3) l’instabilité épigénétique, non totalement indépendante des deux autres mécanismes, associée à une hyperméthylation de certaines régions de l’ADN (CIMP : CpG Island Methylation Phenotype) induisant une inactivation transcriptionnelle de gènes suppresseurs de tumeur (ex. : TP53) (8, 9).
Les microsatellites sont des séquences de l’ADN formées par une répétition de motifs composés de 1 à 6 nucléotides distribuées le long des régions codantes et non codantes du génome (8, 10). Ces séquences sont particulièrement exposées à des erreurs lors de la réplication de l’ADN par la polymérase, avec des insertions/délétions (indel) mono ou di-nucléotidiques. Ces erreurs sont normalement détectées et corrigées par les protéines du système MMR (MisMatch Repair), système biologique hautement conservé de réparation des mésappariements de l’ADN, en charge du maintien de l’intégrité du génome. Les protéines du système MMR comprennent quatre membres : mutL homologue 1 (MLH1), mutS homologue 2 (MSH2), mutS homologue 6 (MSH6) et postmeiotic segregation increased 2 (PMS2) (8, 10). Elles fonctionnent par paires (hétérodimères) associant MLH1 avec PMS2 et MSH2 avec MSH6, l’expression de la seconde étant chaque fois dépendante de la première. Une perte d’expression de ces protéines (MLH1, associée par interdépendance à une perte par dégradation protéolytique de PMS2 ; MSH2, associée à une perte de MSH6 ; perte isolée de PMS2 ou MSH6) induit un défaut de réparation de ces erreurs et l’accumulation de mutations ponctuelles de type indel, entraînant un décalage du cadre de lecture (8, 10). Ces erreurs s’accumulent par milliers dans le génome et en particulier au niveau des microsatellites, dont l’instabilité est le reflet phénotypique de la déficience du système MMR. La perte d’expression d’une protéine du système MMR est la conséquence de l’inactivation biallélique du gène correspondant :
- par mutation constitutionnelle d’un des allèles et perte somatique du second (syndrome de Lynch, ou cancer colique héréditaire sans polypose, associé à un spectre d’autres cancers notamment de l’endomètre, de l’estomac et des voies urinaires ; représentant environ 3 % des CRC et un tiers des cas de CCR MSI) ou beaucoup plus rarement mutation homozygote (syndrome CMMRD),
- ou par extinction épigénétique (hyperméthylation du promoteur du gène)
Les altérations de PMS2, MSH2 et MSH6 sont toujours constitutionnelles. Les extinctions de MLH1 peuvent quant à elles survenir dans un contexte soit constitutionnel soit sporadique, par hyperméthylation du promoteur du gène dans le cadre d’une mutation BRAF V600E (30 % des CCR MSI) ou d’un phénotype hyperméthylateur sans mutation de BRAF.
La recherche d’un phénotype MSI peut être réalisée par deux méthodes validées :
- par IHC sur coupes en paraffine (marquage des quatre protéines MLH1, MSH2, MSH6 et PMS2) : il s’agit alors de rechercher la perte d’expression de protéines du système MMR ;
- par biologie moléculaire après extraction d’ADN selon la technique du Pentaplex® analysant cinq microsatellites (BAT25, BAT26, NR-21, NR-22, NR-24, NR-27) : le phénotype est dit MSI en présence d’une instabilité d’au moins trois de ces microsatellites (11).
À noter qu’aucune des deux techniques n’est parfaitement sensible et ni spécifique. L’IHC peut avoir une sensibilité diminuée selon la méthode de fixation et le protocole d’IHC n’est pas standardisé, pouvant conduire à des erreurs d’interprétation par des pathologistes non experts (11). La PCR présente une sensibilité limitée en cas de faible cellularité tumorale et de contamination importante par de l’ADN de tissu normal (> 90 %), et des faux négatifs peuvent ainsi être observés dans le cadre de biopsies, de tumeurs mucineuses ou presque stérilisées par des traitements néoadjuvants (11). La recherche de mutation de la chaperonne HSP110 (technique HT17) a une sensibilité plus élevée mais est encore en cours de validation (11). Des erreurs diagnostiques (faux positifs) sont possibles soulignant l’utilité de la réalisation des deux techniques pour confirmer le statut MSI en cas de positivité de l’une d’entre elles, en particulier si l’administration d’une immunothérapie est discutée (12).
Le statut des microsatellites doit être systématiquement déterminé par IHC ou PCR chez les patients atteints de CCR pour 3 raisons :
- la recherche d’une prédisposition au cancer colorectal/syndrome de Lynch, en particulier en cas de diagnostic de CCR avant l’âge de 70 ans ou quel que soit l’âge au diagnostic en cas de cancers multiples du spectre du syndrome de Lynch chez un même patient ou chez deux apparentés au premier degré, afin d’orienter le patient vers une consultation d’oncogénétique (impact sur la surveillance du cas index et des apparentés) ;
- au stade localisé : décision thérapeutique de chimiothérapie adjuvante pour les stades II ;
- au stade métastatique : marqueur prédictif de réponse à l’immunothérapie.
Les CCR MSI de stade localisé (II/III, environ 15 % des patients) ont un meilleur pronostic spontané et une moindre sensibilité à la chimiothérapie par fluoropyrimidine (13, 14). L’utilisation d’une fluoropyrimidine en monothérapie n’améliore pas la survie des patients par rapport au traitement chirurgical seul (14). En revanche, l’adjonction de l’oxaliplatine à une fluoropyrimidine permet une amélioration significative de la survie des patients présentant une tumeur de stade III, mais pas de ceux ayant une tumeur de stade II (15, 16). Ainsi la chimiothérapie adjuvante n’est pas indiquée pour les CCR de stade II à haut risque [T4, < 12 ganglions (17)] MSI en raison de leur meilleur pronostic spontané (sauf T4b), et la chimiothérapie adjuvante des stades III doit comporter de l’oxaliplatine (FOLFOX ou XELOX) (18). À l’inverse, chez les patients atteints de CCR de stade IV, le statut MSI, plus rare (environ 5 % des patients), a été associé à des survies plus courtes mais son impact pronostique n’est pas définitivement établi (19-21).
La charge mutationnelle élevée associée au phénotype MSI génère une quantité importante de néoantigènes. Ces antigènes, très différents des protéines du soi, sont fortement immunogènes. Ceci est à l’origine d’infiltrats immunitaires lymphocytaires « Crohn-like » très évocateurs du phénotype MSI (8). De plus, cette immunogénicité rend les tumeurs MSI très sensibles à l’immunothérapie par inhibiteurs des points de contrôle immunitaire (anti-programmed death-1 [PD1] et son ligand [PD-L1] et anti-cytotoxic T-lymphocyte associated protein 4 [CTLA4]) (22). Après plusieurs études de phase II positives avec le pembrolizumab (anti-PD1) ou le nivolumab (anti-PD1) seul ou associé à l’ipilimumab (anti-CTLA4) en situation métastatique (23, 24) et néoadjuvante (25), l’étude de phase III KEYNOTE-177 présentée à l’ASCO 2020 et à l’ESMO 2020 a montré la supériorité de l’immunothérapie par pembrolizumab par rapport à la chimiothérapie de première ligne en terme de SSP (objectif principal : 16,5 vs. 8,2 mois, Hazard ratio [HR] : 0,60, p=0,0002), de taux de réponse (67 % vs. 51 %), de toxicités de grade 3-4 (22 % vs. 66 %) et de qualité de vie chez les patients atteints de CCR métastatique MSI (26). Plusieurs autres études de phase III sont en cours, notamment avec le nivolumab seul ou associé à l’ipilimumab (NCT04008030) (27).
Ces anticorps ont obtenu depuis 2016 une autorisation de mise sur marché (AMM) aux États-Unis « pan-tumeur » de phénotype MSI ; ceci n’a malheureusement pas été suivi rapidement d’une AMM européenne. Les résultats positifs de l’étude KEYNOTE-177 ont permis l’obtention d’une AMM européenne pour le pembrolizumab dans le CCR métastatique en première ligne (avis EMA rendu le 29/01/2021), mais pas encore d’un remboursement en France. Dans l’attente, ces anticorps sont accessibles dans le cadre d’essais cliniques (ou à la charge de l’établissement). En France, l’étude PRODIGE 54 SAMCO (NCT03186326) évalue l’avelumab (anticorps anti-PD-L1) vs. chimiothérapie en traitement de deuxième ligne des CCR métastatiques MSI.
À noter que d’autres altérations moléculaires peuvent être responsables d’un phénotype hypermuté et sensible à l’immunothérapie, en particulier les mutations de la polymérase E pour lesquelles le nivolumab est accessible dans le cadre d’une cohorte ACSé soutenue par l’INCa (NCT03012581) et sont l’objet d’une cohorte COMAD (28).
Mutations RAS
La voie de RAS-RAF-MEK-ERK (mitogen activated protein kinase, MAPK) est une des principales voies de signalisation en aval des récepteurs à activité tyrosine kinase dont le récepteur du facteur de croissance épidermique (EGFR), favorisant la croissance, la différenciation, la prolifération et la survie cellulaire (Figure 1). Au sommet de cette cascade de signalisation, RAS comprend une famille de GTPases cytoplasmiques dont il existe plusieurs isoformes (KRAS, NRAS, HRAS) (29). Une fois activées, elles stimulent en aval plusieurs voies effectrices, dont celle des MAPK, mais aussi de PI3K-AKT-mTOR et de Ral-GEF.
Les anticorps anti-EGFR ont été développés dans le CCR métastatique initialement pour les patients dont la tumeur exprimait ce récepteur [études BOND (30) et NCI-CO17 (31)]. Il avait été supposé que l’activité de ces anticorps serait corrélée au pourcentage de cellules positives pour l’EGFR et/ou à l’intensité d’expression de ce récepteur, mais ceci n’a pas été confirmé dans ces premières études cliniques. En 2006, Lièvre et al. (32)
ont décrit que, plutôt que le statut d’expression de l’EGFR, les mutations activatrices de KRAS, conduisant l’activation constitutive de la voie indépendamment de la liaison du ligand au récepteur, étaient un prédicteur de la résistance au traitement par cetuximab, mAc anti-EGFR, chez les patients atteints de CCR métastatique. Une analyse a posteriori de l’étude NCI-O17 a ensuite été conduite en fonction du statut de mutation KRAS (codons 12 et 13 de l’exon 2) et a montré que le bénéfice clinique était en effet confiné à la population de patients ayant un CCR avec exon 2 de KRAS non muté (33). De plus, la valeur prédictive négative des mutations de KRAS pour le bénéfice d’un traitement anti-EGFR a été confirmée avec le panitumumab, autre mAc anti-EGFR (34, 35). Les études ont ensuite été restreintes aux CCR sans mutation de KRAS exon 2 (présente dans environ 40 % des CCR métastatiques). Outre les mutations de l’exon 2 de KRAS, 10 % à 15 % supplémentaires de patients atteints d’un CCR métastatique sont porteurs d’autres mutations RAS. Il s’agit notamment de mutations de NRAS (codons 12 ou 13 dans l’exon 2, et codons 59 ou 61 dans l’exon 3), et de mutations de KRAS en dehors de l’exon 2 (codons 59 ou 61 dans l’exon 3, et codons 117 ou 146 dans l’exon 4) (36). Chez les patients porteurs de l’une de ces mutations de RAS dans le génome de leur tumeur, plusieurs études, y compris une analyse rétrospective des études CRYSTAL (FOLFIRI ± cetuximab), OPUS (FOLFOX ± cetuximab) et PRIME (FOLFOX ± panitumumab), ont montré que l’ajout de mAc anti-EGFR à divers protocoles de chimiothérapie dans différentes lignes de traitements n’apportait aucun bénéfice, et pouvait même avoir un effet délétère spécifiquement lorsqu’ils étaient associés à une chimiothérapie comportant de l’oxaliplatine (36-38). Cela a été confirmé par Sorich et al.
(39) dans une revue systématique et méta-analyse de neuf essais contrôlés randomisés évaluant le traitement par mAc anti-EGFR chez près de 6 000 patients. À côté de leur valeur prédictive, le rôle pronostique des mutations de RAS chez les patients atteints de CCR reste controversé. Un effet négatif des mutations KRAS a été rapporté à la fois en situation adjuvante et métastatique mais sans conclusion définitive.
Sur la base de ces résultats, les traitements anti-EGFR (cetuximab et panitumumab) ne doivent être prescrits qu’aux patients présentant un statut tumoral de type non muté lors d’une analyse étendue de RAS (exons 2, 3 et 4 de KRAS et NRAS), représentant environ 50 % des patients (AMM modifiée en 2013) (40). Le statut de mutation de RAS peut être obtenu par différentes méthodes, telles que la PCR en temps réel spécifique de mutation, le séquençage de Sanger, le pyroséquençage, la technique BEAMing, le NGS et le séquençage didésoxynucléotidique. Il existe des tests génétiques ciblés commerciaux capables de détecter des mutations de RAS dans des échantillons de CCR, dont des techniques simplifiées (ex. : Idylla®). Les matériaux tissulaires des lésions primitives ou métastatiques peuvent être utilisés pour cette recherche puisque le taux de concordance du statut de mutation entre sites primitifs et métastatiques atteint 93 % (41).

Figure 1 : Voies de signalisation du récepteur de l’EGF (EGFR)
Représentation schématique des principales voies effectrices de l’EGFR : la voie Ras-ERK et ses interactions avec les voies PI3K / AKT / mTOR et RalGEF / Ral et d’autres récepteurs [adapté de Neuzillet et al. (93)]. FdT : facteur de transcription, GAP : protéine activant la GTPase, GDP : guanosine diphosphate, GEF : facteur d’échange nucléotidique de guanine, GTP : guanosine triphosphate.
Mutation BRAF V600E
Plus récemment, d’autres mutations de la voie des MAPK, comme celles du gène BRAF, ont été identifiées dans les CCR. Ces mutations de BRAF sont plus rares (8 % à 10 %) et sont en grande majorité (plus de 90 % des cas) représentées par la mutation activatrice V600E (42). Si la valeur prédictive de la mutation BRAF V600E sur la réponse aux anti-EGFR reste débattue, sa valeur pronostique péjorative est bien établie, en particulier aux stades avancés (SG médiane inférieure à un an) (20) mais aussi pour les stades localisés microsatellites stables (MSS)(43). Les mutations BRAF V600E sont associées à un phénotype particulier : plus fréquemment observées chez la femme âgée, avec des tumeurs du côlon droit, peu différenciées et mucineuses, et des métastases ganglionnaires et péritonéales (44). Ces caractéristiques pronostiques et clinico-pathologiques sont spécifiques à la mutation V600E, et les CCR porteurs d’autres mutations de BRAF (plus rares, environ 2 % des CCR) ont un comportement biologique proche des tumeurs non mutées (45). À noter qu’environ 20 %-40 % des tumeurs mutées BRAF V600E présentent un phénotype MSI sporadique et répondent aussi bien à l’immunothérapie que les tumeurs MSI non BRAF mutées. En raison de leur pronostic sombre avec un accès limité aux traitements de deuxième ligne (<40 %) (46), l’intensification thérapeutique par tri-chimiothérapie (FOLFIRINOX) associée au bevacizumab en première ligne est considérée comme un standard (40), mais sur la base d’un niveau de preuve faible (analyses de sous-groupe) (47) et avec des données récentes contradictoires (pas de différence entre doublet plus bevacizumab vs. triplet plus bevacizumab chez les patients avec CCR métastatique BRAF muté dans une méta-analyse récente) (48). À la différence des mutations BRAF V600E observées dans les mélanomes, les CCR mutés BRAF V600E ne répondent pas à l’inhibition de BRAF en monothérapie, notamment en raison de boucles de rétrocontrôle impliquant l’EGFR, nécessitant un blocage multi-étagé de la voie (42). L’étude de phase III BEACON, a comparé chez des patients atteints de CCR métastatique en deuxième ou troisième ligne, un triplet encorafenib (anti-BRAF), binimetinib (anti-MEK) plus cetuximab (anti-EGFR) ou un doublet encorafenib plus cetuximab vs. une chimiothérapie par FOLFIRI ou irinotécan associée au cetuximab (49). Cette étude a montré la supériorité du triplet ou doublet de thérapies ciblées sur la SG (objectif principal, médiane : 9,0 mois avec le triplet vs. 5,4 mois dans le bras contrôle, HR : 0,52, p<0,001, et 8,4 mois avec le doublet) avec également une amélioration significative de la SSP, du taux de réponse et de la qualité de vie. Ces résultats ont permis l’obtention d’une AMM américaine et européenne (prix et remboursement en cours de discussion) pour le doublet, actuellement accessible en France dans le cadre d’une ATU de cohorte. Les résultats en première ligne avec le triplet (étude de phase II ANCHOR) étaient plus mitigés (SSP médiane de 4,9 mois) (50).
Ainsi, au-delà d’un biomarqueur pronostique, la mutation BRAF V600E est devenue un marqueur prédictif compagnon pour le doublet encorafenib cetuximab. Sur la base de ces résultats, la détermination du statut BRAF est requise chez les patients atteints de CCR métastatique, en particulier pour permettre d’accéder à ce traitement en deuxième ligne.
Biomarqueurs émergents (HER2, MET, NTRK, Immunoscore, CMS)
D’autres altérations moléculaires émergent en tant que marqueurs prédictifs permettant la sélection de patients pour des traitements par thérapies ciblées dans les formes métastatiques. Parmi elles, les amplifications de HER2 (1 %-8 % des CCR métastatiques), associées à la résistance primaire ou acquise sous anti-EGFR, définissent un sous-groupe de patients chez lesquels une activité des anti-HER2 a été rapportée (association trastuzumab plus pertuzumab, trastuzumab plus lapatinib, trastuzumab plus tucatinib, et plus récemment anticorps conjugué trastuzumab-deruxtecan) (51). Elles sont observées le plus souvent en l’absence de mutation KRAS mais les deux altérations peuvent co-exister. Ces altérations peuvent être recherchées par IHC et/ou ISH. Par analogie avec le cancer de l’estomac, le statut HER2 positif est défini comme un score IHC 3+ ou 2+ avec ISH positif (52). D’autres altérations (ex. : mutation/amplification de MET, mutation de PIK3CA, délétion de PTEN…) peuvent également être identifiées, dans les tumeurs naïves ou pré-traitées par anti-EGFR, mais ne font pas l’objet de traitement validé à ce jour (51). Elles peuvent être détectées dans le cadre de panels NGS.
Au même titre que le phénotype MSI, qui peut être observé dans une grande variété de tumeurs de différents primitifs, les réarrangements du gène NTRK (codant pour le récepteur kinase de la tropomyosine [TRK]) sont une altération génétique « agnostique » survenant avec une faible fréquence (<0,5 %) dans une grande variété de cancers, notamment les CCR. TRK est un récepteur exprimé de façon ubiquitaire et les fusions géniques impliquant des gènes NTRK conduisent à la synthèse de protéines chimériques avec une fonction kinase activée ou surexprimée de manière constitutive conférant un potentiel oncogène (53). Le larotrectinib est un inhibiteur de TRK. Il a démontré une activité dans une étude « basket » de phase II ayant inclus une grande variété de tumeurs de l’adulte et de l’enfant avec fusion NTRK, lui ayant permis d’obtenir une AMM américaine et européenne « pan-tumeur » (54). Néanmoins, en France, la Commission de la Transparence a rejeté le remboursement de cette molécule dans l’indication des cancers de l’adulte (avis du 09/07/2020). Aussi, la recommandation du TNCD de recherche de cette altération par IHC confirmée par séquencage (NGS ARN) ou séquençage d’emblée sera probablement révisée. Des études de phase II se poursuivent avec le larotrectinib et l’entrectinib.
Des scores biologiques ont également été proposés dans les CCR localisés. Un Immunoscore de quantification in situ de l’infiltrat lymphocytaire en IHC a été évalué dans le cadre d’un consortium international dans un but de standardisation de ce score en pratique clinique. La reproductibilité du score standardisé a été validée dans une série de près de 2 700 patients atteints d’un cancer du côlon de stade I, II ou III et sa valeur pronostique dans les cancers du côlon localisés a été confirmée (55). L’Immunoscore était un prédicteur fort du risque de récidive, de la SG et de la SSM (tous p<0,0001), indépendamment de l’âge du patient, du sexe, du statut MSI et d’autres facteurs pronostiques existants. Cependant, son rôle dans la prédiction des bénéfices de la chimiothérapie est incertain et des preuves solides de son rôle pronostique dans un ensemble de données de stade II seulement font actuellement défaut.
Plusieurs classifications moléculaires des CCR ont fait l’objet d’un consensus international pour retenir quatre sous-groupes transcriptomiques (signatures ARN d’expression génique) reliés à des paramètres cliniques et moléculaires et à des données de survie (Consensus Molecular Subtypes – CMS1 : « immun » avec MSI et activation immunitaire ; CMS2 : « canonique » avec différenciation épithéliale et activation de la voie WNT/MYC ; CMS3 : « métabolique » avec dérégulation épithéliale et métabolique ; CMS4 : « mésenchymateux » avec activation de la voie TGFb, angiogenèse et implication du stroma) (56). D’autres signatures géniques sont devenues des candidats potentiels pour la stratification pronostique dans les CCR localisés. L’Oncotype DX (57) et le GeneFx Colon (58) ont été validés dans l’analyse multivariée de cohortes randomisées prospectives indépendantes de cancers du côlon de stade II. Leur utilisation est limitée en raison du manque de valeur prédictive du bénéfice de la chimiothérapie et des faibles marges de différenciation pronostique entre les scores élevés, intermédiaires et faibles.
L’intérêt potentiel de ces paramètres biologiques pronostiques n’est pas validé en pratique vis-à-vis de l’impact qu’ils pourraient avoir sur la décision thérapeutique (17, 18).
Place de l’ADN tumoral circulant
Les mutations de RAS et de BRAF ainsi que d’autres mutations peuvent être détectées à partir de l’ADN tumoral circulant (ADNtc), aussi qualifié de « biopsies liquides », avec une bonne concordance du statut mutationnel tissulaire et plasmatique. Des études ont montré l’intérêt de la détection d’ADNtc en termes de stratification pronostique des patients, en particulier en situation adjuvante, et de monitoring thérapeutique (suivi des clones résistants) chez les patients métastatiques traités par anti-EGFR (59). Néanmoins, la détection d’ADNtc a une sensibilité limitée en l’absence de métastases hépatiques et les techniques d’analyse ne sont pas standardisées. Elle est en cours d’évaluation dans le cadre d’essais cliniques et n’est pas encore validée en routine (17, 18).
Cancer œsogastrique
Surexpression/amplification HER2
HER2 est un autre récepteur à activité tyrosine kinase membre de la famille de l’EGFR (ERBB) (1). Il n’a pas de ligand connu et est le partenaire d’hétérodimérisation préféré des autres récepteurs ERBB (ex. : HER3). Dans le cancer gastrique, HER2 agit comme un oncogène. La surexpression d’HER2 est associée à une amplification de segments du chromosome 17. HER2 est surexprimé dans environ 30 % des cancers gastriques de type intestinal, 15 % de type mixte, et environ 5 % de type diffus (60). Selon la localisation de la tumeur, la surexpression d’HER2 est plus fréquente au niveau du cardia / jonction œso-gastrique (JOG) (environ 30 %) que de l’estomac (20 %) (60).
La surexpression de HER2 chez les patients atteints d’un cancer gastrique avancé a été corrélée à un mauvais pronostic et à une maladie plus agressive, bien que ces études soient limitées par leurs faibles effectifs, l’hétérogénéité des patients, des méthodes de tests HER2 et de classification du statut HER2 (61).
Cliniquement, HER2 a été établi comme biomarqueur prédictif pour les thérapies ciblées anti-HER2. La preuve de concept de l’inhibition de HER2 a été obtenue pour la première fois dans le cancer du sein HER2-positif, à la fois dans les situations de maladie précoce et métastatique avec des agents ciblant ce récepteur, tels que les mAc trastuzumab, pertuzumab, trastuzumab-emtansine (T-DM1), et IMK comme le lapatinib (1). Dans les cancers gastriques et de la JOG avancés, la surexpression d’HER2 est prédictive de réponse au trastuzumab, établie sur la base de l’étude de phase III ToGA (62). Cette étude avait montré la supériorité de l’ajout du trastuzumab à la chimiothérapie par fluoropyrimidine et cisplatine par rapport à la chimiothérapie seule chez les patients atteints de cancers gastriques ou de la JOG avancés HER2-positifs en termes de SG (objectif principal, médiane : 13,8 vs. 11,1 mois, HR : 0,74, p=0,0046) et de SSP, et a permis au trastuzumab d’obtenir une AMM dans cette indication en 2010. En revanche, à la différence du cancer du sein, le pertuzumab, le T-DM1 et le lapatinib n’ont pas montré d’efficacité dans les cancers de l’estomac et de la JOG avancés HER2-positifs (1). Le trastuzumab-deruxtecan a quant à lui montré des premiers résultats d’efficacité récents (63). Le statut HER2 n’a en revanche pas d’impact sur la décision thérapeutique dans les formes localisées, le niveau de preuve pour l’ajout de thérapies anti-HER2 dans cette indication étant insuffisant.
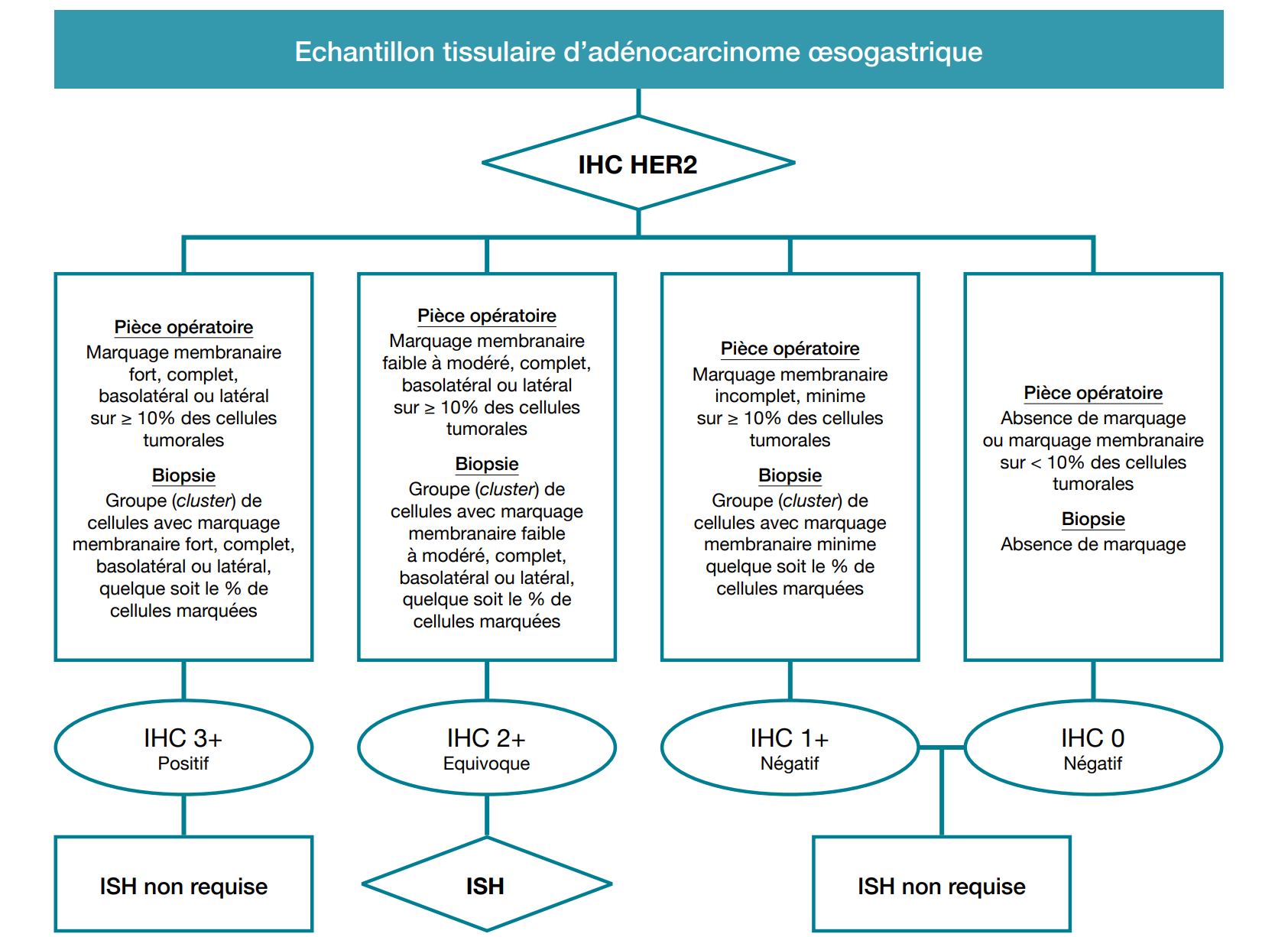
La détermination du statut HER2 doit être réalisée chez tous les patients avec un adénocarcinome gastrique, de la JOG ou de l’œsophage de stade avancé, éligibles à un traitement systémique, et elle est recommandée (mais non obligatoire) chez tous les patients opérés d’un cancer gastrique (TNCD, 2018). L’IHC doit être la première technique réalisée (64). L’évaluation de l’expression d’HER2 en IHC dans le cancer de l’estomac fait l’objet d’un protocole spécifique, distinct de celui utilisé pour le cancer du sein, afin de prendre en compte les spécificités de l’expression d’HER2 dans l’estomac, notamment son hétérogénéité intra-tumorale et la présence d’un marquage souvent incomplet et localisé aux membranes baso-latérales et latérales (Figure 2) (65, 66). Des échantillons chirurgicaux représentatifs ou un nombre suffisant de biopsies (au moins huit) sont nécessaires (64). L’ISH est indiquée en cas de marquage IHC 2+ (64). Au moins 20 cellules sans chevauchement doivent être évaluables.
L’amplification HER2 est définie par un ratio HER2/chromosome 17 ≥ 2.0. Une tumeur est classée HER2-positive en cas de score IHC 3+ ou IHC 2+ avec ISH positive (64). Une bonne concordance a été rapportée entre la tumeur primitive et les métastases, ainsi qu’entre la biopsie et les échantillons de tumeurs réséquées, bien qu’une diminution de l’expression de HER2 ait été observée après chimiothérapie néoadjuvante (67, 68).

Figure 2 : Algorithme de scoring HER2 dans le cancer gastrique [adapté de Bartley et al. (64)]
MSI et biomarqueurs émergents (EBV, MSI, PD-L1)
Des classifications moléculaires ont permis de démembrer les cancers gastriques en quatre sous types : les tumeurs positives pour le virus Epstein- Barr (EBV), qui présentent des mutations récurrentes de PIK3CA, une hyperméthylation extrême de l’ADN et une amplification de JAK2, PD-L1 et PD-L2 ; les tumeurs MSI, qui présentent des taux de mutation élevés, y compris des mutations de gènes codant pour des protéines de signalisation oncogéniques ciblables ; des tumeurs génomiquement stables, qui sont enrichies en type histologique diffus et porteuses d’altérations génétiques impliquant des protéines activant la GTPase de la famille Rho ; et les tumeurs avec instabilité chromosomique, qui présentent une aneuploïdie marquée et une amplification focale des récepteurs tyrosine kinases (69).
Environ 10 % des cancers de l’estomac ont un phénotype MSI (70). À la différence du CCR, les méthodes de diagnostic du statut MSI dans les tumeurs gastriques (et plus largement autres tumeurs non-colorectales) sont moins bien standardisées et il est recommandé de réaliser la technique complémentaire pour confirmer si l’une des deux techniques (IHC ou PCR) est positive (11). Outre la recherche d’un syndrome de Lynch, la détermination du statut MSI dans l’adénocarcinome gastrique a deux intérêts : aide à la décision pour le traitement péri-opératoire et sélection des patients pour une immunothérapie. Les tumeurs gastriques MSI sont associées à un meilleur pronostic et sont davantage chimiorésistantes. Aux stades localisés, l’analyse a posteriori de ce sous-groupe de patients de l’étude MAGIC a montré que les patients avec tumeur MSI non traités par chimiothérapie avaient un excellent pronostic (médiane de SG non atteinte) alors que les patients MSI traités par chimiothérapie péri-opératoire avaient une SG particulièrement limitée (9 mois de médiane), suggérant un potentiel effet délétère de la chimiothérapie (71). De même, les données d’une étude asiatique ont montré l’absence d’efficacité de la chimiothérapie adjuvante par XELOX dans le sous-groupe des patients MSI (72). La chimiothérapie péri-opératoire par FLOT (ou FOLFOX pour les patients plus fragiles), standard dans les cancers gastriques opérables, ne semble donc pas la stratégie la plus adaptée en cas de tumeur MSI (73). En revanche, au même titre que les CCR MSI, ces tumeurs sont sensibles à l’immunothérapie (taux de réponse au pembrolizumab : 86 %), en particulier aux stades avancés (74). En situation péri-opératoire, l’étude GERCOR NEONIPIGA (NCT04006262), permet à ces patients d’avoir un accès à une immunothérapie par nivolumab (anti-PD1) associé à l’ipilimumab (anti-CTLA4). Les cancers gastriques EBV positifs semblent aussi être de bons répondeurs à l’immunothérapie (taux de réponse au pembrolizumab : 100 %), mais ces données reposent sur des études de faibles effectifs et demandent à être confirmées pour recommander la détermination du statut EBV (détection par sonde EBER en ISH) en routine (74).
En dehors du phénotype MSI, l’expression de PD-L1 est aussi prédictive de réponse à l’immunothérapie. Dans le cancer gastrique, elle est évaluée par un score combiné d’expression dit « CPS », qui correspond au ratio du nombre de cellules tumorales ou stromales PD-L1-positives sur le nombre de cellules tumorales, multiplié par 100. Des études antérieures en ligne tardive avaient montré une activité plus importante des anti-PD1 chez les patients dont les tumeurs exprimaient plus fortement le PD-L1 (phase III KEYNOTE-062, seuil CPS ≥ 10) (74, 75). L’étude de phase III Checkmate-649 a évalué l’ajout du pembrolizumab à la chimiothérapie de première ligne dans l’adénocarcinome de l’estomac et de la JOG et a confirmé une activité de ces traitements plus particulièrement pour les patients avec expression forte de PD-L1 (score CPS ≥ 5, population cible de l’objectif principal de l’étude). L’étude était positive à la fois sur la SG (médiane : 14,4 vs. 11,1 mois, HR : 0,71, p< 0,0001) et la SSP (7,7 vs. 6 mois, HR : 0,68, p< 0,0001) (76). L’AMM de l’immunothérapie dans l’estomac pourrait être limitée à ce sous-groupe de patients.
Cancer du pancréas : mutations germinales BRCA
Les cancers du pancréas sont caractérisés par une fréquence très élevée (> 90 %) de mutations activatrices KRAS, non ciblables à ce jour en
thérapeutique. Les thérapies ciblées administrées à des patients atteints de cancers du pancréas non sélectionnés sur le plan moléculaire ont, pendant plus d’une décennie, toutes échoué à améliorer significativement la survie des patients (1). Des progrès récents sont venus de l’identification de petits sous-groupes de patients avec des altérations moléculaires spécifiques.
Environ 10 % des adénocarcinomes du pancréas sont développés dans un contexte de prédisposition génétique. L’altération génétique constitutionnelle la plus fréquente est la mutation de BRCA2, ou moins souvent de BRCA1. Ces gènes sont impliqués dans les syndromes de prédisposition aux cancers du sein, de l’ovaire et de la prostate (mode de transmission autosomique dominant, comme le syndrome de Lynch), et peuvent aussi s’associer dans certaines familles à des cancers du pancréas. Elles sont plus fréquentes dans certains groupes ethniques, en particulier chez les Juifs ashkénazes (14 % vs. 7 %). BRCA est impliqué dans la réparation des cassures double brin de l’ADN par recombinaison homologue (77). Lorsqu’il est inactivé dans les cellules tumorales, elles deviennent déficientes pour réparer ces cassures, les rendant particulièrement sensibles aux chimiothérapies induisant des cassures de l’ADN comme les platines (78). De plus, le système poly(ADP-ribose) polymérase (PARP) prend alors partiellement le relais pour réparer les cassures de l’ADN et devient le « talon d’Achille » des cellules tumorales. L’enzyme PARP répare les cassures simple brin de l’ADN et les inhibiteurs de PARP bloquent de manière catalytique cette enzyme. Des cassures double brin se produisent alors pendant la réplication de l’ADN et les cellules tumorales dépourvues de système BRCA ne peuvent les réparer et meurent par le principe de létalité synthétique (77). Ce concept avait déjà été démontré dans les cancers du sein, de l’ovaire ou de la prostate. L’efficacité de l’olaparib (inhibiteur
de PARP) a été évaluée dans l’essai de phase III POLO en monothérapie comme traitement de maintenance chez les patients atteints de cancer du pancréas métastatique et porteurs d’une mutation germinale de BRCA1 ou BRCA2, dont la maladie était contrôlée après au moins quatre mois d’une chimiothérapie de première ligne comportant un platine (79). L’étude était positive sur son objectif principal, la SSP (médiane : 7,4 vs. 3,8 mois, HR : 0,53, p= 0,004), et ces résultats ont conduit à l’obtention d’une AMM européenne dans cette indication.
Les altérations germinales de BRCA sont recherchées au niveau sanguin, dans le cadre d’une consultation d’oncogénétique ou non. Ses conditions de réalisation font actuellement l’objet d’un groupe de travail à l’INCa afin d’établir des recommandations. Il n’est pas possible à ce jour d’étendre l’indication de l’olaparib aux mutations somatiques (c’est-à-dire détectées uniquement dans la tumeur) et aux mutations autres que BRCA1/2 induisant un phénotype similaire de déficience du système de réparation de l’ADN par recombinaison homologue (HRD ou « BRCAness ») (80).
L’étude PRODIGE MAZEPPA en cours (NCT04348045) établit un profil moléculaire tumoral des patients ayant un adénocarcinome du pancréas métastatique contrôlé après quatre mois (8 cycles) de chimiothérapie d’induction par FOLFIRINOX, et comprend un bras permettant l’administration d’un traitement de maintenance par olaparib chez les patients présentant une BRCAness somatique.
À côté des altérations de BRCA, d’autres altérations peuvent être accessibles à des traitements ciblés : phénotype MSI (1 % des patients, recommandé pour discuter l’administration d’une immunothérapie dans le cadre d’essais cliniques) et transcrits de fusion (ex. : NTRK, NRG1, ALK, RET ; qui peuvent être recherchés par des panels NGS ARN ciblés type Archer®), plus particulièrement chez les patients non porteurs de mutations KRAS (5 %-10 % des patients, à discuter mais non recommandé en routine) (80, 81).
Cancers des voies biliaires : place du séquençage NGS
Comme pour le cancer du pancréas, les essais avec des thérapies ciblées « classiques » (principalement anti-EGFR et anti-angiogéniques) dans des populations de patients non sélectionnés sur le plan moléculaire, seuls ou en association avec la chimiothérapie, n’ont démontré aucun bénéfice de survie significatif chez les patients atteints de cancers des voies biliaires (CVB) avancés (1).
Ces dernières années, les connaissances sur l’hétérogénéité moléculaire des CVB ont considérablement progressé avec l’avènement des analyses génomiques et transcriptomiques par NGS, ouvrant de nouvelles voies pour les thérapies ciblées et la stratification thérapeutique des patients. Certaines anomalies moléculaires ont été identifiées spécifiquement selon le type de CVB (cholangiocarcinomes [CCA] intra-hépatiques, extra- hépatiques, adénocarcinome de la vésicule biliaire) (82).
L’essai MOSCATO-01 a fourni les premières preuves qu’un profil moléculaire était faisable et pouvait apporter un bénéfice chez ces patients (83). Le taux de réussite pour détecter au moins une altération moléculaire ciblable était d’environ 70 %. L’administration de traitements ciblés sur ces anomalies a montré un bénéfice clinique (défini par le rapport SSP avec la thérapie ciblée sur SSP avec la ligne antérieure > 1,3) chez 80 % des patients et un taux de réponse objective de 33 %.
Les principales altérations ciblables décrites sont (i) dans les CCA intra-hépatiques, des mutations de l’isocitrate déshydrogénase-1 (IDH1), impliquée dans le métabolisme cellulaire mitochondrial, et des réarrangements du récepteur au facteur de croissance des fibroblastes (FGFR2), présents chacun chez 10 % à 20 % des patients ; (ii) des altérations des gènes de la famille de l’EGFR (mutation ou amplification HER2, HER3) dans les CCA extra-hépatiques et les cancers de la vésicule biliaire ; (iii) d’autres transcrits de fusions (notamment, NTRK) et des mutations BRAF ; (iv) une instabilité des microsatellites, présente chez 5 %-10 % des CVB (84). Les altérations d’IDH1 (étude de phase III positive avec l’ivosidenib (85) chez les patients pré-traités) et de FGFR2 [études de phase II positives avec le pemigatinib (86) et l’infigratinib (87) chez les patients pré-traités et phase III en cours en première ligne] sont les deux principales cibles thérapeutiques « modernes » dans les CCA intra-hépatiques dont le développement clinique est le plus avancé (84).
Des procédures d’accès à ces molécules par ATU sont en cours d’activation et des essais cliniques les évaluent en première ligne. Ces opportunités thérapeutiques et les essais thérapeutiques en cours incitent à réaliser précocement au cours du parcours une détermination du statut MSI et HER2 en IHC (simples et peu coûteux) et un panel NGS ADN (pour la recherche des mutations, incluant IDH1, BRAF, et la famille EGFR/HER) et ARN (pour la recherche de transcrits de fusion, incluant FGFR2 et NTRK). L’ESMO a récemment validé cette recommandation de réalisation d’un panel NGS chez les patients atteints de CVB en routine (7). L’étude PRODIGE SAFIR ABC 10 débutera prochainement, et proposera un traitement de maintenance bioguidé à partir du profil ADN/ARN de la tumeur après une chimiothérapie d’induction par gemcitabine et cisplatine.
Tumeurs stromales gastro-intestinales (GIST) : KIT et PDGFRA
L’imatinib, un IMK dirigé contre l’activité kinase BCR-ABL, c-KIT et PDGFRA, a été le premier agent ciblé approuvé dans le traitement des tumeurs stromales gastro-intestinales (GIST) avancées (2). L’imatinib a été initialement développé en 1998 pour la leucémie myéloïde chronique, une maladie provoquée par la kinase BCR-ABL avec un phénotype de dépendance oncogénique, réalisant, historiquement, la première preuve de concept de l’inhibition spécifique d’une activité tyrosine kinase en tant qu’intervention thérapeutique cliniquement utile, et donnant le premier succès remarquable des IMK en onco-hématologie. Sur la base de son large spectre d’inhibition du kinome s’étendant à c-KIT, et sachant depuis la fin des années 1990 que les GIST étaient entraînées par des mutations de gain de fonction de ce récepteur tyrosine kinase, l’imatinib a été testé chez un seul patient avec une GIST métastatique chimioréfractaire et a montré une efficacité remarquable, avec une réponse métabolique complète et prolongée.
Suite à la publication de ce cas clinique en 2001, un essai ouvert de phase II a montré l’efficacité de l’imatinib (400 mg ou 600 mg par jour) chez les patients atteints de GIST avancée c-KIT-positive (88). Ces résultats ont permis l’obtention d’une AMM en 2002. Par la suite, il a été démontré que les tumeurs répondaient à des degrés variables en fonction du type de mutation KIT portée par la GIST. Au-delà des mutations de KIT, d’autres profils moléculaires de GIST ont été décrits (mutation de PDGFRA, GIST « wild-type » sans mutation de KIT ni PDGFRA).
Ces résultats ont été le point de départ de la stratification génotypique des patients dans la prise en charge des GIST (2). Dans les GIST résécables, le génotype est un outil complémentaire à la classification de Miettinen pour évaluer le risque de récidive (89). Les GIST avec mutation de KIT ont un risque de récidive supérieur à celles avec mutation de PDGFRA, les GIST wild-type ayant un risque intermédiaire entre ces 2 groupes (89). Au stade métastatique, le génotypage prédit la sensibilité à l’imatinib. Les mutations de KIT sont les plus fréquentes dans l’exon 11 (63 %-66 %), suivi de l’exon 9 (6 %-13 %), et sont rarement retrouvées dans les exons 13 et 17 (< 2 %) (90, 91). La plupart des GIST avec mutations de KIT exon 11 ou 13 sont sensibles à l’imatinib ; la dose de 400 mg/jour en première intention est validée (90-92). Les mutations de KIT exon 9 sont associées à la localisation de l’intestin grêle ou du côlon, au phénotype à cellules fusiformes avec des caractéristiques clinico-pathologiquement agressives et à une moindre sensibilité à l’imatinib ; l’utilisation d’imatinib à forte dose (800 mg/jour) dans les formes avancées est soutenue par l’augmentation de la SSP médiane, mais pas en SG, qui a été observée dans la méta-analyse MetaGIST (90-92). Les mutations de KIT exon 17 sont rares et certaines d’entre elles (par exemple, D816V) sont résistantes à l’imatinib (90, 91). Les mutations de PDGFRA (10 %-15 %) sont fréquentes dans les GIST gastriques, avec un phénotype épithélioïde et un comportement indolent (90, 91). Les mutations des exons 12 et 14 sont peu fréquentes mais présentent une sensibilité à l’imatinib (90, 91). À l’inverse, la mutation prédominante de PDGFRA, l’exon 18 D842V, est associée à une résistance à l’imatinib, ainsi qu’au sunitinib et au régorafénib ; elles peuvent être ciblées par des molécules spécifiques (avapritinib, accessible en ATU de cohorte). Les GIST wild-type (négatives pour les mutations de KIT et PDGFRA, 10 %-15 %) sont de génotype hétérogène et peuvent inclure des mutations dans HRAS, NRAS, BRAF, NF1 ou le complexe SDH ; certaines d’entre elles sont liées à des syndromes familiaux (90, 91). Elles ont une sensibilité variable à l’imatinib mais cette molécule ne doit pas être exclue (90-92). L’ensemble de ces résultats justifie la réalisation d’un profil mutationnel systématique au diagnostic de GIST (89).
Conclusion
Au cours des dernières années, l’oncologie digestive a connu des évolutions avec plusieurs études de phase III positives ayant permis de positionner de nouvelles molécules sur des populations de patients sélectionnés. Le développement de ces nouveaux traitements a été indissociable de l’identification de marqueurs compagnons pour en orienter la prescription ; cette meilleure sélection des patients a permis d’obtenir des résultats dans des indications où les thérapies ciblées « pour tous » avaient échoué (l’exemple le plus marquant étant celui des CVB). Les altérations moléculaires ESCAT I/II dans les différentes localisations sont résumées dans le tableau 2 (7). Malgré ces résultats positifs et leur niveau de preuve, ces innovations ne peuvent malheureusement pas être transposées immédiatement en pratique clinique en France du fait de problématiques d’AMM ou de remboursement (ex. : inhibiteurs de NTRK). Une réflexion sur l’accélération et la diffusion de l’accès à l’innovation, prenant en compte tout à la fois le bénéfice clinique, la dimension éthique (dans le cas de l’impossibilité de prescription d’un traitement efficace), et les implications en termes de coûts liés aux traitements eux-mêmes et aux recherches moléculaires associées doit être engagée.
| Altération moléculaire | Prévalence | Molécules | Classe ESCAT |
|---|
| Cancer colorectal métastatique | Mutations KRAS
Mutations NRAS | 44 %
4 % | Cetuximab, panitumumab | Non applicable |
| Mutation BRAFV600E | 8,5 % | Encorafenib plus cetuximab | IA |
| Instabilité des microsatellites (MSI) | 4 %-5 % | Pembrolizumab | IA |
| Fusions NRTK | 0,5 % | Larotrectinib, entrectinib | IC |
| Amplification ERBB2 (HER2) | 2 % | Trastuzumab plus pertuzumab, trastuzumab plus lapatinib | IIB |
| Cancer gastrique métastatique | Amplification ERBB2 (HER2) | 16 % | Trastuzumab | IA |
| Instabilité des microsatellites (MSI) | 8 % | Pembrolizumab, nivolumab | IC |
| Fusions NTRK | 2 % | Larotrectinib | IC |
| Amplification EGFR | 6 % | Cetuximab, ABT-806 | IIB |
| Amplification MET | 3 % | Crizotinib | IIB |
| Cancer du pancréas métastatique | Mutations germinales BRCA1/2 | 1 %-4 % | Olaparib | IA |
| Instabilité des microsatellites (MSI) | 1 %-3 % | Pembrolizumab | IC |
| Fusions NTRK | < 1 % | Larotrectinib, entrectinib | IC |
| Cholangiocarcinomes avancés | Mutations IDH1 | 20 % | Ivosidenib | IA |
| Fusions FGFR2 | 15 % | Pemigatinib | IB |
| Instabilité des microsatellites (MSI) | 2 % | Pembrolizumab | IC |
| Fusions NTRK | 2 % | Larotrectinib, entrectinib | IC |
| Mutation BRAFV600E | 5 % | Dabrafenib plus trametinib | IIB |
Tableau 2 : Principales altérations ESCAT I/II en oncologie digestive [d’après Mosele et al. (7)]
Références
- Neuzillet C, Rousseau B, Kocher H, et al. Unravelling the pharmacologic opportunities and future directions for targeted therapies in gastro-intestinal cancers Part 1: GI carcinomas. Pharmacol Ther 2017; 174: 145-172.
- Neuzillet C, de Mestier L, Rousseau B, et al. Unravelling the pharmacologic opportunities and future directions for targeted therapies in gastro- intestinal cancers part 2: Neuroendocrine tumours, hepatocellular carcinoma, and gastro-intestinal stromal Pharmacol Ther 2018; 181: 49-75.
- Biomarkers and surrogate endpoints: preferred definitions and conceptual Clin Pharmacol Ther 2001; 69: 89-95.
- Febbo PG, Ladanyi M, Aldape KD, et al. NCCN Task Force report: Evaluating the clinical utility of tumor markers in J Natl Compr Canc Netw 2011; 9 Suppl 5: S1-32; quiz S33.
- Test compagnon associé à une thérapie ciblée : définitions et méthode d’évaluation. Guide méthodologique HAS – Février
- Mateo J, Chakravarty D, Dienstmann R, et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO Scale
- Mosele F, Remon J, Mateo J, et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a
- Vilar E, Gruber Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol 2010; 7: 153-162.
- Laurent-Puig P, Agostini J, Maley K. [Colorectal oncogenesis]. Bull Cancer 2010; 97: 1311-1321.
- Collura A, Lefevre JH, Svrcek M, et al. [Microsatellite instability and cancer: from genomic instability to personalized medicine]. Med Sci (Paris) 2019; 35: 535-543.
- Svrcek M, Lascols O, Cohen R, et al. MSI/MMR-deficient tumor diagnosis: Which standard for screening and for diagnosis? Diagnostic modalities for the colon and other sites: Differences between Bull Cancer 2019; 106: 119-128.
- Cohen R, Hain E, Buhard O, et al. Association of Primary Resistance to Immune Checkpoint Inhibitors in Metastatic Colorectal Cancer With Misdiagnosis of Microsatellite Instability or Mismatch Repair Deficiency Status. JAMA Oncol 2019; 5: 551-555.
- Dienstmann R, Mason MJ, Sinicrope FA, et Prediction of overall survival in stage II and III colon cancer beyond TNM system: a retrospective, pooled biomarker study. Ann Oncol 2017; 28: 1023-1031.
- Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol 2010; 28: 3219-3226.
- Andre T, de Gramont A, Vernerey D, et al. Adjuvant Fluorouracil, Leucovorin, and Oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF Mutation and Mismatch Repair Status of the MOSAIC Study. J Clin Oncol 2015; 33: 4176-4187.
- Tougeron D, Mouillet G, Trouilloud I, et al. Efficacy of Adjuvant Chemotherapy in Colon Cancer With Microsatellite Instability: A Large Multicenter AGEO Study. J Natl Cancer Inst 2016; 108.
- Argiles G, Tabernero J, Labianca R, et al. Localised colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2020; 31: 1291-1305.
- Lecomte T, André T, Bibeau F, et al. Cancer du côlon non métastatique. Thésaurus National de Cancérologie Digestive, Janvier 2019.
- Innocenti F, Ou FS, Qu X, et al. Mutational Analysis of Patients With Colorectal Cancer in CALGB/SWOG 80405 Identifies New Roles of Microsatellite Instability and Tumor Mutational Burden for Patient J Clin Oncol 2019; 37: 1217-1227.
- Venderbosch S, Nagtegaal ID, Maughan TS, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS Clin Cancer Res 2014; 20: 5322-5330.
- Taieb J, Shi Q, Pederson L, et al. Prognosis of microsatellite instability and/or mismatch repair deficiency stage III colon cancer patients after disease recurrence following adjuvant treatment: results of an ACCENT pooled analysis of seven Ann Oncol 2019; 30: 1466-1471.
- Cohen R, Rousseau B, Vidal J, et al. Immune Checkpoint Inhibition in Colorectal Cancer: Microsatellite Instability and Target Oncol 2020; 15: 11-24.
- Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 Science 2017; 357: 409-413.
- Overman MJ, Lonardi S, Wong KYM, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/ Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol 2018; 36: 773-779.
- Chalabi M, Fanchi LF, Dijkstra KK, et Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med 2020; 26: 566-576.
- Andre T, Shiu KK, Kim TW et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal N Engl J Med 2020; 383: 2207-2218.
- Overman MJ, Ernstoff MS, Morse Where We Stand With Immunotherapy in Colorectal Cancer: Deficient Mismatch Repair, Proficient Mismatch Repair, and Toxicity Management. Am Soc Clin Oncol Educ Book 2018; 38: 239-247.
- Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330-337.
- Neuzillet C, Tijeras-Raballand A, de Mestier L, et al. MEK in cancer and cancer therapy. Pharmacol Ther 2014; 141: 160-171.
- Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 2004; 351: 337-345.
- Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal N Engl J Med 2007; 357: 2040-2048.
- Lievre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal Cancer Res 2006; 66: 3992-3995.
- Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal N Engl J Med 2008; 359: 1757-1765.
- Van Cutsem E, Peeters M, Siena S, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol 2007; 25: 1658-1664.
- Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal J Clin Oncol 2008; 26: 1626-1634.
- Peeters M, Douillard JY, Van Cutsem E, et al. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J Clin Oncol 2013; 31: 759-765.
- Fakih Metastatic colorectal cancer: current state and future directions. J Clin Oncol 2015; 33: 1809-1824.
- Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016.
- Sorich MJ, Wiese MD, Rowland A, et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015; 26: 13-21.
- Phelip JM, Tougeron D, Leonard D, et Metastatic colorectal cancer (mCRC): French intergroup clinical practice guidelines for diagnosis, treatments and follow-up (SNFGE, FFCD, GERCOR, UNICANCER, SFCD, SFED, SFRO, SFR). Dig Liver Dis 2019; 51: 1357-1363.
- Wong NA, Gonzalez D, Salto-Tellez M, et al. RAS testing of colorectal carcinoma-a guidance document from the Association of Clinical Pathologists Molecular Pathology and Diagnostics Group. J Clin Pathol 2014; 67: 751-757.
- Lievre A, de la Fouchardiere C, Samalin E, et al. [BRAF V600E-mutant colorectal cancers: Where are we?]. Bull Cancer 2020; 107: 881-895.
- Taieb J, Le Malicot K, Shi Q, et Prognostic Value of BRAF and KRAS Mutations in MSI and MSS Stage III Colon Cancer. J Natl Cancer Inst 2017; 109.
- de la Fouchardiere C, Cohen R, Malka D, et al. Characteristics of BRAF (V600E) Mutant, Deficient Mismatch Repair/Proficient Mismatch Repair, Metastatic Colorectal Cancer: A Multicenter Series of 287 Oncologist 2019; 24: e1331-e1340.
- Jones JC, Renfro LA, Al-Shamsi HO, et al. (Non-V600) BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J Clin Oncol 2017; 35: 2624-2630.
- Seligmann JF, Fisher D, Smith CG, et al. Investigating the poor outcomes of BRAF-mutant advanced colorectal cancer: analysis from 2530 patients in randomised clinical trials. Ann Oncol 2017; 28: 562-568.
- Cremolini C, Loupakis F, Antoniotti C, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE Lancet Oncol 2015; 16: 1306-1315.
- Cremolini C, Antoniotti C, Stein A, et al. Individual Patient Data Meta-Analysis of FOLFOXIRI Plus Bevacizumab Versus Doublets Plus Bevacizumab as Initial Therapy of Unresectable Metastatic Colorectal Cancer. J Clin Oncol 2020;
- Kopetz S, Grothey A, Yaeger R, et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N Engl J Med 2019; 381: 1632-1643.
- Grothey A, Tabernero J, Taieb J, et al. LBA-5 ANCHOR CRC: a single-arm, phase 2 study of encorafenib, binimetinib plus cetuximab in previously untreated BRAF V600E-mutant metastatic colorectal Annals of Oncology 2020; 31: S242-S243.
- Cohen R, Pudlarz T, Delattre JF, et al. Molecular Targets for the Treatment of Metastatic Colorectal Cancers (Basel) 2020; 12.
- Valtorta E, Martino C, Sartore-Bianchi A, et al. Assessment of a HER2 scoring system for colorectal cancer: results from a validation study. Mod Pathol 2015; 28: 1481-1491.
- Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016; 1: e000023.
- Drilon A, Laetsch TW, Kummar S, et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med 2018; 378: 731-739.
- Pages F, Mlecnik B, Marliot F, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 2018; 391: 2128-2139.
- Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal Nat Med 2015; 21: 1350-1356.
- Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011; 29: 4611-4619.
- Niedzwiecki D, Frankel WL, Venook AP, et al. Association Between Results of a Gene Expression Signature Assay and Recurrence-Free Interval in Patients With Stage II Colon Cancer in Cancer and Leukemia Group B 9581 (Alliance). J Clin Oncol 2016; 34: 3047-3053.
- Bach S, Sluiter NR, Beagan JJ, et Circulating Tumor DNA Analysis: Clinical Implications for Colorectal Cancer Patients. A Systematic Review. JNCI Cancer Spectr 2019; 3: pkz042.
- Van Cutsem E, Bang YJ, Feng-Yi F, et al. HER2 screening data from ToGA: targeting HER2 in gastric and gastroesophageal junction Gastric Cancer 2015; 18: 476-484.
- Chen C, Yang JM, Hu TT, et al. Prognostic role of human epidermal growth factor receptor in gastric cancer: a systematic review and meta-analysis. Arch Med Res 2013; 44: 380-389.
- Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled Lancet 2010; 376: 687-697.
- Shitara K, Bang YJ, Iwasa S, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric N Engl J Med 2020; 382: 2419-2430.
- Bartley AN, Washington MK, Colasacco C, et al. HER2 Testing and Clinical Decision Making in Gastroesophageal Adenocarcinoma: Guideline From the College of American Pathologists, American Society for Clinical Pathology, and the American Society of Clinical Oncology. J Clin Oncol 2017; 35: 446-464.
- Ruschoff J, Dietel M, Baretton G, et al. HER2 diagnostics in gastric cancer-guideline validation and development of standardized immunohistochemical testing. Virchows Arch 2010; 457: 299-307.
- Stenzinger A, von Winterfeld M, Aulmann S, et al. Quantitative analysis of diagnostic guidelines for HER2-status assessment. J Mol Diagn 2012; 14: 199-205.
- Bozzetti C, Negri FV, Lagrasta CA, et Comparison of HER2 status in primary and paired metastatic sites of gastric carcinoma. Br J Cancer 2011; 104: 1372-1376.
- Watson S, Validire P, Cervera P, et al. Combined HER2 analysis of biopsies and surgical specimens to optimize detection of trastuzumab- eligible patients in eso-gastric adenocarcinoma: a GERCOR Ann Oncol 2013; 24: 3035-3039.
- Cancer Genome Atlas Research Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014; 513: 202-209.
- Polom K, Marano L, Marrelli D, et al. Meta-analysis of microsatellite instability in relation to clinicopathological characteristics and overall survival in gastric Br J Surg 2018; 105: 159-167.
- Smyth EC, Wotherspoon A, Peckitt C, et al. Mismatch Repair Deficiency, Microsatellite Instability, and Survival: An Exploratory Analysis of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) JAMA Oncol 2017; 3: 1197-1203.
- Choi YY, Kim H, Shin SJ, et al. Microsatellite Instability and Programmed Cell Death-Ligand 1 Expression in Stage II/III Gastric Cancer: Post Hoc Analysis of the CLASSIC Randomized Controlled study. Ann Surg 2019; 270: 309-316.
- Zaanan A, Bouche O, Benhaim L, et al. Gastric cancer: French intergroup clinical practice guidelines for diagnosis, treatments and follow-up (SNFGE, FFCD, GERCOR, UNICANCER, SFCD, SFED, SFRO). Dig Liver Dis 2018; 50: 768-779.
- Kim ST, Cristescu R, Bass AJ, et Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med 2018; 24: 1449-1458.
- Shitara K, Van Cutsem E, Bang YJ, et al. Efficacy and Safety of Pembrolizumab or Pembrolizumab Plus Chemotherapy vs Chemotherapy Alone for Patients With First-line, Advanced Gastric Cancer: The KEYNOTE-062 Phase 3 Randomized Clinical JAMA Oncol 2020.
- Moehler M, Shitara K, Garrido M, et al. LBA6_PR Nivolumab (nivo) plus chemotherapy (chemo) versus chemo as first-line (1L) treatment for advanced gastric cancer/gastroesophageal junction cancer (GC/GEJC)/esophageal adenocarcinoma (EAC): First results of the CheckMate 649 Annals of Oncology 2020; 31: S1191.
- Gourley C, Balmana J, Ledermann JA, et al. Moving From Poly (ADP-Ribose) Polymerase Inhibition to Targeting DNA Repair and DNA Damage Response in Cancer J Clin Oncol 2019; 37: 2257-2269.
- Patch AM, Christie EL, Etemadmoghadam D, et al. Whole-genome characterization of chemoresistant ovarian Nature 2015; 521: 489-494.
- Golan T, Hammel P, Reni M, et Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med 2019; 381: 317-327.
- Singhi AD, George B, Greenbowe JR, et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Gastroenterology 2019; 156: 2242-2253 e2244.
- Pishvaian MJ, Blais EM, Brody JR, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecula profiling: a retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol 2020; 21: 508-518.
- Valle JW, Lamarca A, Goyal L, et al. New Horizons for Precision Medicine in Biliary Tract Cancer Discov 2017; 7: 943-962.
- Verlingue L, Malka D, Allorant A, et al. Precision medicine for patients with advanced biliary tract cancers: An effective strategy within the prospective MOSCATO-01 Eur J Cancer 2017; 87: 122-130.
- Vienot A, Neuzillet C. Continuum of care for advanced biliary tract Clin Res Hepatol Gastroenterol 2020.
- Abou-Alfa GK, Macarulla T, Javle MM, et Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 2020; 21: 796-807.
- Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 Lancet Oncol 2020; 21: 671-684.
- Javle M, Lowery M, Shroff RT, et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced J Clin Oncol 2018; 36: 276-282.
- Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal N Engl J Med 2002; 347: 472-480.
- Landi B, Blay JY, Bonvalot S, et al. Gastrointestinal stromal tumours (GISTs): French Intergroup Clinical Practice Guidelines for diagnosis, treatments and follow-up (SNFGE, FFCD, GERCOR, UNICANCER, SFCD, SFED, SFRO). Dig Liver Dis 2019; 51: 1223-1231.
- Blay JY, Casali PG, Dei Tos AP, et al. Management of Gastrointestinal Stromal Tumour: Current Practices and Visions for the Future. Oncology 2015; 89: 1-13.
- Nishida T, Blay JY, Hirota S, et al. The standard diagnosis, treatment, and follow-up of gastrointestinal stromal tumors based on Gastric Cancer 2016; 19: 3-14.
- Group Gastrointestinal stromal tumours: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014; 25 Suppl 3: iii21-26.
- Neuzillet C, Hammel P, Tijeras-Raballand A, et al. Targeting the Ras-ERK pathway in pancreatic adenocarcinoma. Cancer Metastasis Rev 2013; 32: 147-162.