Lien d’intérêts
Aucun
Mots-clés
Kystes biliaires ; polykystose hépatique ; maladie de Caroli ; Hamartomes ; Tumeurs mucineuses kystiques.
Abréviations
- ACE : Antigène carcino-embryonnaire
- ALG 8 : Alpha-1,3-Glucosyltransferase
- AMPc : Adénosine monophosphate cyclique
- APC : adenomatous polyposis coli
- AUDC : Acide Ursodésoxycholique
- AUROC : Area Under the Receiver Operating Characteristics curve
- Ca 19-9 : Cancer antigen 19-9
- Ca 72-4 : Cancer antigen 72-4
- CK : cytokératine
- CPRE : Cholangiopancréatographie rétrograde endoscopique
- FDG : Fluorodésoxyglucose
- GANAB : neutral alpha-glucosidase AB
- GGT : gamma-glutamyl transpeptidase
- GIST : Tumeurs stromales gastro-intestinales
- GluIIβ : Glucosidase IIβ
- IGF-1 : insulin growth factor 1
- IRM : imagerie par résonnance magnétique
- LRP5 : Low-density lipoprotein receptor-related protein 5
- mTOR : mechanistic target of rapamycin
- OMS : Organisation Mondiale de la Santé
- PC1 : polycystine 1
- PC2 : polycystine 2
- PET-scan : tomographie à émission de positrons
- PKD1 : gène codant pour la polycystine 1
- PKD2 : gène codant pour la polycystine 2
- PKH : polykystose hépatique
- PKHAD : polykystose hépatique autosomique dominante
- PKHD1 : gène codant pour la fibrocystine
- PKRAD : polykystose rénale autosomique dominante
- PKRAR : polykystose rénale autosomique récessive
- PRKCSH : Protein Kinase C Substrate 80K-H
- PTEN : Phosphatase and tensin homolog
- SEC63/SEC61B : Gènes codant pour des protéines transmembranaires du réticulum endoplasmique
- TIPS : Transjugular Intrahepatic Portosystemic Shunt
- TMK : Tumeurs mucineuses kystiques
- TPI : Tumeurs Papillaires Intracanalaires
- UH : Unités Houndsfield
- VEGF : Vascular Endothelial Growth Factor
Introduction
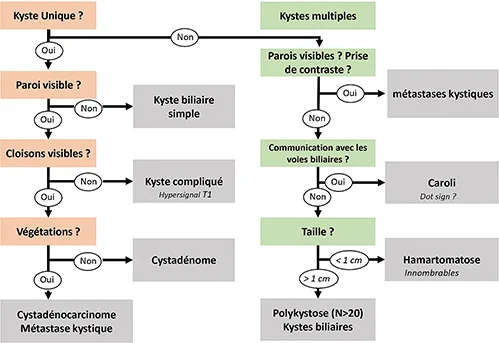
Les kystes hépatiques sont fréquemment rencontrés en pratique clinique quotidienne. Ils rassemblent un groupe de lésions hétérogènes qui varient en fonction de leur pathogenèse, leur présentation clinique, leur aspect en imagerie, et leur gravité potentielle. Les kystes hépatiques bénins sont heureusement les plus fréquents, mais d’autres lésions, parfois difficiles à distinguer d’un kyste simple, peuvent être malignes, voire engager le pronostic vital. Plusieurs classifications des kystes hépatiques sont disponibles dans la littérature ; la plus répandue sépare les kystes hépatiques d’origine infectieuse et non infectieuse et, par extension, les kystes d’origine parasitaire et non parasitaire du foie, car les abcès à pyogènes, habituellement faciles à diagnostiquer du fait de leur présentation clinique et morphologique, sont souvent exclus des algorithmes diagnostiques. Cette mise au point concerne uniquement les lésions kystiques non parasitaires. Pour chacune d’entre elles sont communiquées les hypothèses physiopathologiques, les données épidémiologiques, l’histoire naturelle (en particulier le risque de transformation maligne), le traitement et, bien sûr les clés du diagnostic radiologique. Ces dernières sont résumées et simplifiées, dans la figure 1, qui concerne les lésions fréquemment évoquées dans les discussions diagnostiques. En dehors des kystes biliaires simples, qui dans leur présentation typique, peuvent être diagnostiqués par une simple échographie abdominale, toutes les autres lésions nécessitent la réalisation d’un scanner ou d’une imagerie par résonance magnétique (IRM), soit pour leur caractérisation (ce qui nécessite alors toujours une injection de produit de contraste), soit pour l’estimation volumétrique du parenchyme hépatique épargné (cas de la polykystose hépatique). Dans certaines situations diagnostiques ambiguës, ou pour guider la discussion thérapeutique, d’autres outils peuvent être utiles comme l’échographie de contraste, le PET-scan, la ponction du liquide intra kystique, ou la biopsie, et méritent d’être évoqués. Un résumé des connaissances sur le diagnostic génétique des polykystoses et son futur intérêt clinique est aussi inclus dans cette mise au point.

Figure 1 : Aide-mémoire simplifié d’orientation diagnostique en imagerie devant la découverte d’un kyste hépatique.
Seules les principales lésions kystiques sont répertoriées.
Kystes biliaires
Les kystes biliaires simples sont actuellement les plus fréquentes des formations kystiques hépatiques. Ils peuvent être localisés partout dans le foie. L’appellation de « kyste biliaire », justifiée par la présence d’un épithélium analogue à celui des canaux biliaires, est toujours la plus utilisée, bien que jugée inopportune compte tenu de l’absence de communication de ces kystes avec les voies biliaires. L’appellation « kyste simple », proposée dès les années 1980, bien que plus adaptée pour bien les distinguer des authentiques dilatations kystiques des voies biliaires du syndrome de Caroli et de la fibrose hépatique congénitale (1), reste peu utilisée en France.
Caractéristiques anatomopathologiques
Le kyste biliaire se définit comme une cavité unique, contenant un liquide séreux, limpide ou citrin, pauvre en éléments cellulaires, et ne communiquant pas avec les voies biliaires intrahépatiques (2). Il est bordé d’un épithélium cubique ou cylindrique analogue à celui des canaux biliaires. La composition du liquide intracavitaire est proche de celle de la fraction de secrétion biliaire indépendante des sels biliaires : même concentration hydroélectrolytique, faible concentration en protéines, présence d’IgA sécrétoires et de gamma GT à des taux supérieurs aux concentrations sériques. Il n’existe aucun transfert de grosse molécule entre le sang et le liquide intrakystique. Ces caractéristiques du liquide intrakystique permettent de distinguer les kystes simples des kystes biliaires communicants et des néoplasies kystiques. Fait remarquable, le liquide des kystes biliaires est riche en ACE dans 66 % des cas et riche en Ca19-9 dans 88 % des cas (3).
Histoire naturelle
L’origine des kystes biliaires n’est pas clairement élucidée. Elle est probablement congénitale, et l’hypothèse la plus communément admise est la dilatation kystique d’une voie biliaire aberrante, ou d’un microhamartome (cf. infra). Des microhamartomes ont en effet été décrits associés aux kystes biliaires dans des séries autopsiques, alors fréquemment retrouvés dans la paroi des kystes ou à distance, dans les espaces portes. Ces microhamartomes sont constitués d’un sac épithélial en continuité avec les travées hépatocytaires, mais sans communication canalaire.
L’histoire naturelle des kystes simples reste imparfaitement connue. Cette méconnaissance est influencée par les circonstances de diagnostic (le plus souvent fortuites) et la recommandation, justifiée, de ne pas entreprendre de suivi morphologique chez les patients atteints de kystes biliaires. Avant le développement des explorations morphologiques actuelles, les kystes simples du foie étaient considérés comme rares. En fait, Leur prévalence est de 2 à 5 % dans les séries échographiques (1,4) et peut même atteindre 18 % dans les séries tomodensitométriques (5), probablement parce que le scanner permet de mieux distinguer des kystes de petite taille ou de localisation plus difficile à visualiser en échographie. Certaines données concernant les kystes simples semblent toutefois admises : leur caractère exceptionnel avant l’âge de 10 ans, leur fréquence croissante avec l’âge avec un pic compris entre 50 et 60 ans, et la prédominance féminine (sex ratio : 1,5 à 4/1), encore plus marquée pour les kystes volumineux et symptomatiques (1). Dans 30 % des cas, on retrouve plusieurs kystes et plus rarement de multiples kystes qui peuvent faire évoquer le diagnostic de polykystose hépatique, voire de polykystose hépato-rénale, lorsque les reins sont le siège de quelques kystes banaux. Il n’existe aucune localisation préférentielle. Les kystes découverts à l’échographie mesurent en général moins de 3 cm. Ils peuvent exceptionnellement être de taille spectaculaire, jusqu’à 40 cm, ou contenir plus de 10 litres de liquide. Les kystes de grande taille sont plus susceptibles d’être symptomatiques ou de se compliquer. Au cours du temps, les kystes biliaires augmentent de taille dans moins de 20 % des cas. Les facteurs susceptibles d’influencer le développement des kystes restent mal connus. Ils pourraient impliquer la sécrétine, qui stimule la sécrétion des canaux biliaires, en lien avec une influence hormonale suggérée par la nette prédominance féminine, similaire à ce qui est clairement établi au cours de la polykystose hépatique (cf. infra). Le mécanisme n’a toutefois pas été élucidé.
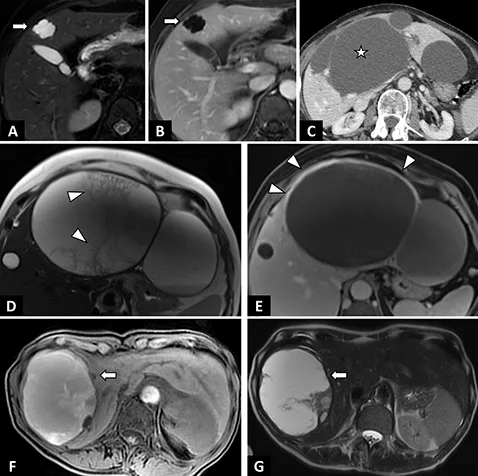
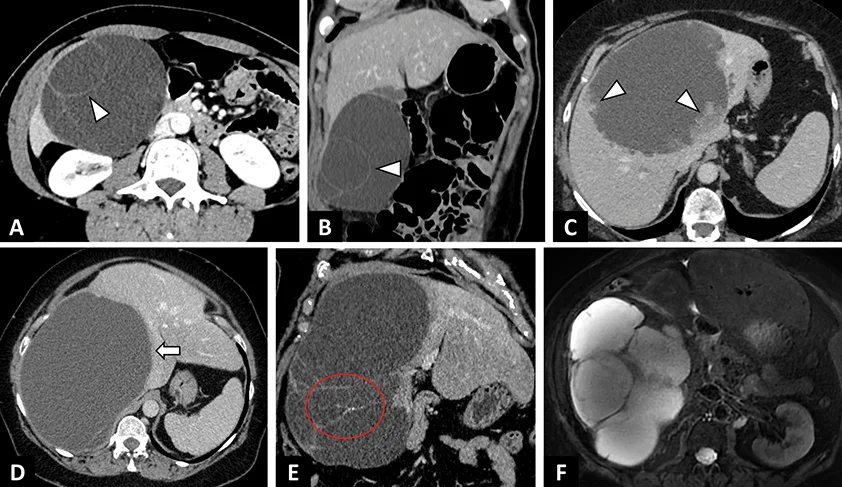
Figure 2 : Exemples de kystes biliaires simples et compliqués |
A–C : Kyste biliaire simple. A. IRM en séquence axiale T2 avec saturation de la graisse. Kyste biliaire en hypersignal T2 liquidien de contours polylobés (flèche).
B. IRM en séquence axiale T1 après injection de gadolinium au temps portal. Absence de rehaussement pariétal (flèche).
C. Scanner abdominal après injection de contraste au temps portal montrant des lésions hypodenses kystiques (étoile), bien limitées, sans paroi du segment IV et du lobe gauche.
D–E : Kyste biliaire infecté chez une patiente de 63 ans révélé par des douleurs fébriles de l’hypochondre droit.
Aspects IRM. D. IRM en séquences axiales T2. Lésion kystique du segment IV à paroi épaisse et de contenu hétérogène (tête de flèche).
E. IRM en séquence axiale T1 après injection de gadolinium au temps portal. Prise de contraste de la paroi du kyste biliaire en rapport avec l’infection kystique.
F–G : Kyste biliaire hémorragique chez une patiente de 76 ans révélé par des douleurs de l’hypochondre droit.
Aspects IRM. F. IRM en séquence axiale T1 avec saturation de la graisse. Lésion kystique du foie droit en hypersignal T1 spontané en rapport avec l’hémorragie.
G. IRM en séquence axiales T2 montrant un contenu hétérogène du kyste hépatique en rapport avec l’hémorragie. (P. Calame, CHU Besançon) |
Diagnostic morphologique
Dans la très grande majorité des cas, les kystes biliaires sont asymptomatiques et de découverte fortuite. L’aspect échographique correspond à une zone anéchogène, ronde, à limite nette sans paroi individualisable, avec un renforcement postérieur des échos. Les signes négatifs importants pour le diagnostic sont l’absence de cloisonnement interne, d’image endokystique et de calcification, même si ces éléments peuvent être exceptionnellement présents, produisant alors des situations trompeuses (tableau 1). En dehors de ces situations, de complications ou de contexte particulier, l’échographie représente le seul examen morphologique nécessaire et suffisant pour poser le diagnostic (1). Au scanner (figure 2C), les kystes biliaires sont des lésions arrondies, hypodenses, sans modification de densité (20 UH) après injection de produit de contraste. En cas de stéatose hépatique marquée, les kystes biliaires peuvent être difficiles à individualiser (6). En IRM (figures 2A,2B), le kyste biliaire est homogène, hypointense en T1 et fortement hyperintense en T2, comme toutes les lésions liquidiennes.
Tableau 1: Principales situations pièges rencontrées dans l’examen morphologique des kystes biliaires| Images pièges | Explications possibles | Éléments rétablissant le diagnostic |
|---|
| image d’allure solide dans le kyste ou sur sa paroi | 1 Hémorragie intrakystique
2 Infection du kyste
3 Rupture pariétale | 1 Sédiment déclive (écho) hyperintense en T1 (IRM)
2 Epaississement pariétal / liquide trouble à la ponction
3 Epanchement péri-hépatique ; élément serpigineux intrakystique
(TDM) mobile (echo) correspondant à la paroi rompue |
|---|
| Présence de cloisons (aspect évocateur de cystadénome), kyste d’allure remaniée | 1 Plusieurs kystes accolés
2 Remaniements hémorragiques
3 Remaniements post sclérose | 1 Absence d’épaississement pariétal, de végétation, de prise
de contraste des parois ou des cloisons, de dilatation biliaire péri-lésionnelle, d’activité au PET scan. Présence d’indentation des parois en regard des cloisons
2 Idem + Signal hyper T1 de la/des pseudo-cloisons
3 Anamnèse |
|---|
| Calcification pariétale | 1 Remaniement
post hémorragique ? | 1 Absence d’épaississement pariétal, de périkyste, de membrane proligère ou de vésicules filles évoquant un kyste hydatique
1 Absence de bourgeon charnu ou de réhaussement après injection évoquant une lésion néoplasique
1 Absence de cloison évoquant un cystadénome |
|---|
Complications
Les complications sont rares et concernent préférentiellement les kystes de grande taille (6). On distingue, par ordre décroissant de fréquence :
- La gêne douloureuse provoquée par un kyste volumineux mettant en tension la capsule de Glisson. Une ponction évacuatrice échoguidée peut avoir un intérêt pour imputer les douleurs au kyste lorsqu’elle entraîne une disparition des douleurs.
- les compressions des organes de voisinage (fréquence : 3 à 9 %) : digestive (gastroduodénale), vasculaire (pouvant être responsable d’un syndrome cave inférieur ou d’une hypertension portale), rénale, ou biliaire. La compression de la convergence biliaire par un volumineux kyste centro-hépatique, entraînant un ictère cholestatique réversible après évacuation du kyste, parfois révélée par une angiocholite, est la compression mécanique la plus fréquente.
- l’hémorragie intrakystique (fréquence : 2 à 5 % ; figures 2F, 2G). Elle est révélée par une douleur de l’hypochondre droit et une augmentation du volume kystique. À l’échographie, il existe un contenu hétérogène et mobile au sein du kyste et, parfois, lors de la lyse du caillot, des cloisons et des végétations peuvent être visibles. Le diagnostic différentiel entre le kyste biliaire hémorragique et le cystadénome est alors particulièrement difficile (4). Il nécessite la réalisation d’une IRM qui visualisera un hypersignal en T1 affirmant la composante hématique (figure 2F), un signal T2 hétérogène (figure 2G) ou simplement plus intense que celui des autres kystes biliaires non compliqués éventuellement présents, et surtout l’absence de prise de contraste de la paroi et des cloisons.
- l’infection bactérienne (1 % des cas ; figures 2D, 2E). Elle peut être iatrogène ou spontanée. Sa présentation clinique et morphologique mime celle d’un abcès hépatique et justifie sans doute la recherche d’une porte d’entrée digestive lorsque une entérobactérie est en cause (cas le plus fréquent).
- Les ruptures pariétales sont exceptionnelles et brutales. Elles peuvent se faire dans la cavité péritonéale, les voies biliaires ou le duodénum, et sont volontiers provoquées par une hémorragie intrakystique. Une rupture intrapéritonéale survenant chez une femme sous contraception œstroprogestative peut faire évoquer à tort le diagnostic de rupture d’adénome hépatique hémorragique.
Traitement
Les kystes biliaires non compliqués ne justifient aucun traitement ni aucun suivi morphologique. L’intérêt d’un suivi morphologique ou d’un traitement préventif des kystes biliaires de grande taille n’a, en particulier, jamais été démontré (2).
La simple ponction évacuatrice a surtout un intérêt diagnostique, son effet n’étant que temporaire du fait de la persistance de l’épithélium sécrétant du kyste. Elle reste essentielle, isolée ou associée à un drainage écho-guidé, dans le traitement des kystes infectés. Dans les autres situations, les deux traitements utilisés sont le traitement radiologique interventionnel percutané, associant l’aspiration du liquide kystique à l’injection d’un produit sclérosant (éthanol, tétracycline, polidocanol ou sérum salé hypertonique), ou le traitement chirurgical (2). La sclérothérapie vise à tarir la sécrétion du kyste par destruction de l’épithélium cavitaire. Après aspiration complète du liquide, le produit sclérosant (le plus souvent l’alcool à 95°) est injecté et laissé en place 20 minutes avant d’être réaspiré (7). Cette méthode a longtemps été considérée comme contre-indiquée en cas de kystes hémorragiques, avant qu’une étude française n’affirme son innocuité et son efficacité comparables en présence ou non d’un contingent hémorragique intra-kystique (8). Les deux inconvénients de cette méthode sont le risque de douleurs lors de l’injection, et de récidive des symptômes à distance du traitement. Une revue systématique récente de 588 kystes biliaires symptomatiques traités par sclérothérapie chez 526 patients avec un suivi médian de 20 mois a documenté une diminution des symptômes dans plus de 72 % des cas et leur disparition complète dans plus de 50 % des cas, ce qui était interprété comme un excellent résultat (7). Dans une méta-analyse de données individuelles rassemblant 86 patients, le seul facteur prédictif d’efficacité à long terme sur les symptômes en analyse multivariée était la réduction du volume du kyste traité mesurée à 6 mois : en cas de disparition des symptômes, la réduction du diamètre à 6 mois était de 79 %, versus 63 % en cas de diminution et 50 % en cas de persistance des symptômes (9).
Le traitement chirurgical consiste en une fenestration ou marsupialisation du kyste au sein de la cavité péritonéale (10). Elle consiste en la résection chirurgicale de la partie saillante du kyste, dont le fond est recouvert de séreuse. La chirurgie laparoscopique est privilégiée, sauf pour les kystes de localisation postérieure, profonde, ou les kystes compliqués du fait des remaniements de la paroi kystique. La kystectomie totale est justifiée en cas de doute diagnostique avec un cystadénome. Une résection hépatique réglée est exceptionnellement nécessaire et réservée à la découverte d’une tumeur intrakystique, aux hémorragies difficiles à contrôler, aux kystes centro-hépatiques, ou aux volumineux kystes du lobe gauche (11).
Après fenestration, la persistance ou la récidive de symptômes ne sont pas rares et ne seraient pas moins fréquentes que ce qui est observé après sclérothérapie, contrairement à ce qui a été rapporté par certains (10). C’est ce que suggère une revue systématique récente rapportant le suivi à 2 ans de 736 patients, dont 471 traités chirurgicalement. La persistance des symptômes était observée dans 3,5 % des cas après sclérothérapie, 2,1 % des cas après chirurgie laparoscopique, et 4,2 % après chirurgie conventionnelle. Compte tenu d’une morbidité plus élevée en cas de chirurgie comparée au traitement percutané, les auteurs recommandaient de réserver la chirurgie aux échecs du traitement percutané (12). Cette recommandation restera toutefois discutable tant qu’aucune randomisée comparant ces deux stratégies thérapeutiques n’aura été réalisée. On ne peut en effet éliminer l’influence d’un biais de publication dans les données actuellement disponibles dans la littérature.
Hamartomes
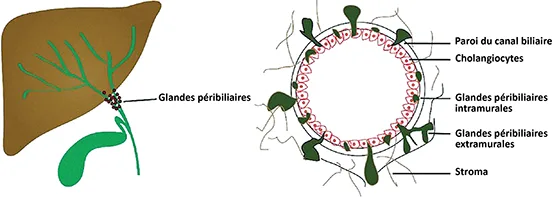
Les hamartomes, également appelés complexes de Von Meyenburg, hamartomes biliaires ou microhamartomes biliaires, sont de petits nodules non encapsulés (généralement < 5 mm), bien circonscrits, constitués de voies biliaires de forme irrégulière, souvent dilatées, bordées par un épithélium cubique bas, enchâssé dans un stroma collagène dense (13). On pense que les hamartomes sont la conséquence d’un remodelage interrompu de la plaque ductale au cours de la phase tardive, lorsque se produit le développement des voies biliaires intrahépatiques de petite taille, et qu’ils sont généralement adjacents aux zones portales. Les hamartomes biliaires constituent la variante la plus fréquente de la maladie hépatique fibropolykystique, survenant chez environ 0,6 % à 2,8 % de la population générale. Les hamartomes peuvent être associés à des kystes biliaires simples, une polykystose rénale, une fibrose hépatique congénitale et/ ou une Maladie de Caroli. Cliniquement, cette affection bénigne reste asymptomatique au cours de la vie et ne nécessite aucune prise en charge ni confirmation histologique chez les patients présentant une présentation d’imagerie typique.
Imagerie
Les hamartomes sont fréquemment découverts de façon fortuite lors d’examens d’imagerie réalisés pour d’autres raisons. Ils se présentent le plus souvent sous la forme de petites lésions kystiques (moins de 1 cm), nettement délimitées et de taille globalement uniforme, dispersées dans tout le parenchyme hépatique. Ils sont le plus souvent très nombreux. On parle alors d’hamartomatose. À l’examen échographique, les petits hamartomes sont généralement échogènes. Les hamartomes millimétriques ne peuvent pas être identifiés comme tels et sont généralement interprétés comme une échostructure hépatique hétérogène diffuse. Les hamartomes supracentimétriques peuvent apparaître hypoéchogènes ou anéchogènes avec une image « en queue de comète » qui correspond à un artéfact de réverbération des petits kystes et peuvent être interprétées à tort comme des calculs intrahépatiques. Au scanner, les complexes de Von Meyenburg apparaissent comme de multiples lésions hypodenses, qui ne se réhaussent pas après injection de produit de contraste. L’IRM (figures 3D, 3E, 3F), met en évidence des lésions clairement délimitées qui apparaissent hypointenses en T1 et nettement hyperintenses en T2, formant alors un aspect caractéristique de « ciel étoilé » ou de « tempête de neige » en T2. Les voies biliaires intra- et extrahépatiques ne sont généralement pas dilatées et ne communiquent pas avec les kystes (6).
Les hamartomes peuvent être difficiles à caractériser en raison de leur petite taille. Le diagnostic différentiel avec des métastases hépatiques multiples de petite taille est rarement résolu à l’échographie seule, ce qui justifie le recours à une imagerie en coupe, où la taille uniforme des kystes et l’absence de toute prise de contraste lésionnelle rétabliront facilement le diagnostic. On a toutefois décrit la possibilité d’un réhaussement nodulaire mural partiel après injection de contraste. Cela dépend de la nature du stroma péri-kystique et de la présence d’une branche porte anormale. En cas de contexte carcinologique, un recours à la biopsie, bien que de rentabilité médiocre compte tenu de la petite taille des lésions, peut alors être tenté, et un suivi morphologique indispensable (14).
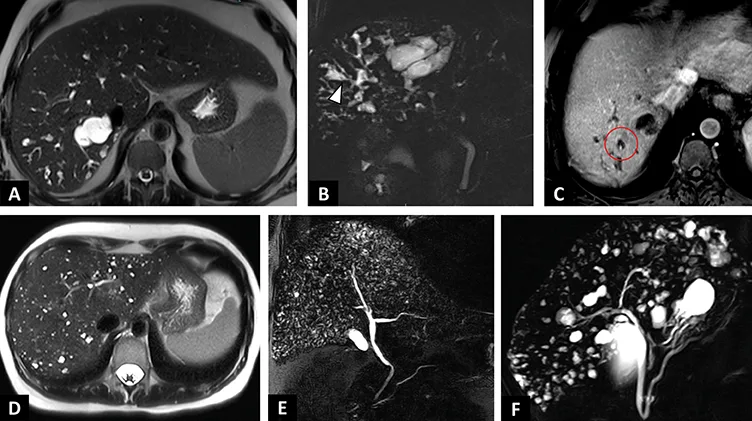
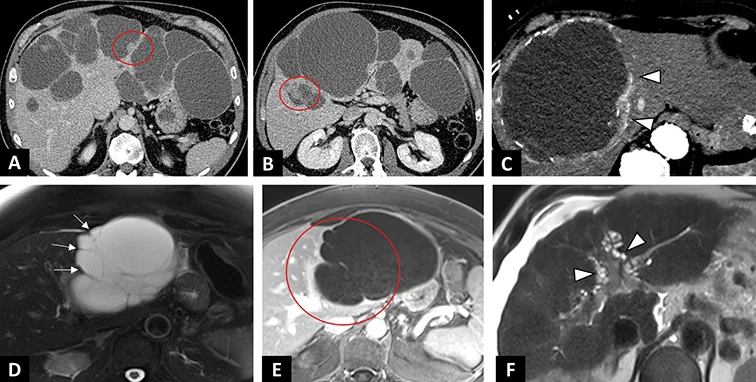
Figure 3 : Anomalies de la plaque ductale |
| A–C : Syndrome de Caroli chez un patient de 47 ans découvert à l’occasion d’un épisode d’angiocholite. A. IRM en séquence axiale T2 montrant des lésions kystiques hépatiques en hypersignal T2 liquidien, communiquant avec les voies biliaires. B. Séquences de cholangioIRM en 2D radiaires. Dilatations kystiques des voies biliaires en doigt de gants (tête de flèche) sans sténose à l’origine des dilatations. C. IRM en séquence axiale T1 après injection de gadolinium au temps portal. « dot sign » (cercle rouge) correspondant à la visualisation d’un vaisseaux porte au sein d’un kyste hépatique. |
| D–F : Exemples d’hamartomatoses biliaires. D. IRM en séquence axiale T2. Innombrables lésions kystiques en hypersignal T2 infracentimé triques de l’ensemble des segments hépatiques. E. Séquences de cholangioIRM en 2D radiaires. Innombrables lésions kystiques hépatiques millimétriques réalisant un aspect en « ciel étoilé ». F. Séquences de cholangioIRM en 2D radiaires. Multiples lésions kystiques hépatiques de plus grosse taille en rapport avec des hamartomes biliaires. |
Histoire naturelle
Les complications des hamartomes sont extrêmement rares. Un seul cas de transplantation hépatique pour des angiocholites à répétition attribuées, peut-être à tort, à des hamartomes a été rapporté (15) et la survenue de complications liées à la fibrose hépatique congénitale possiblement associée n’a pas été spécifiquement décrite chez les patients atteints d’hamartomes. Les hamartomes sont généralement considérés comme bénins, mais plusieurs cas cliniques ont suggéré qu’il existait un risque de transformation en cholangiocarcinome (13). Certaines observations histologiques des pièces d’exerèse, particulièrement convaincantes, ont décrit la transition sans perte de continuité entre l’épithélium bénin, une transformation épithéliale hyperplasique, dysplasique, et une progression adjacente vers un carcinome invasif (16,17). Ce phénomène a été corroboré par des analyses moléculaires documentant la présence des mêmes mutations entre le cholangiocarcinome et les hamartomes (18). Les changements génétiques moléculaires spécifiques pour la transformation des hamartomes en cholangiocarcinome n’ont pas été définitivement établis. Contrairement aux adénomes des voies biliaires, les mutations BRAF observées dans certains cholangiocarcinomes intrahépatiques ne sont pas présentes dans les complexes de Von Meyenburg (13). Des études récentes ont suggéré que des changements génétiques séquentiels étaient nécessaires à la transformation des hamartomes bénins en cholangiocarcinome invasif. Dans l’étude portant sur deux patients atteints d’hamartomatose associée à un cholangiocarcinome qui a fourni la première démonstration moléculaire de l’association entre ces deux lésions, la perte d’hétérozygotie au niveau de 20 loci clés avait été examinée au sein du cholangiocarcinome, des hamartomes éloignés de la tumeur, et des lésions intermédiaires où était observée la transition de l’hamartome vers le cholangiocarcinome. Pour certains loci, une même perte d’hétérozygotie était ainsi observée dans les hamartomes et les cholangiocarcinomes. Elle affectait les oncogènes p16, p53, APC et PTEN, bien connus pour être impliqués dans le développement du cholangiocarcinome (18).
Ces données histologiques et moléculaires récentes justifient en pratique clinique la surveillance morphologique régulière des patients atteints d’hamartomes. À la lumière de ces données, la survenue d’un ictère, d’une augmentation de l’alpha fœtoprotéine, ou d’une image intrakystique chez un patient porteur d’hamartomes doit faire craindre l’apparition d’un cholangiocarcinome et poser l’indication d’une résection chirurgicale (19).
Maladie de Caroli
La maladie de Caroli est une maladie congénitale bénigne, caractérisée par une dilatation kystique segmentaire uni ou bilobaire des voies biliaires intrahépatiques. Sa description a été faite en 1958 par Jacques Caroli. Son incidence est estimée à 1/1 000 000. Elle touche autant les hommes que les femmes, et plus de 80 % des cas sont diagnostiqués avant l’âge de 30 ans.
Il existe deux formes de la maladie, l’une associée à une fibrose hépatique congénitale, aussi appelée syndrome de Caroli, et l’autre, simple, survenant seule. La forme simple serait moins fréquente et associée à une atteinte plus focalisée. La maladie de Caroli s’accompagne d’une incidence accrue de lithiase biliaire, d’angiocholites et d’abcès du foie. L’absence de cirrhose lors du diagnostic est habituelle. Une hypertension portale secondaire à la fibrose hépatique congénitale peut en revanche déjà être présente. La maladie de Caroli peut être associée à plusieurs atteintes
rénales : polykystose rénale récessive de l’enfant, rein éponge, et maladie de Cacchi et Ricci (1).
Origine et histoire naturelle
La maladie de Caroli est une maladie génétique dont le mode de transmission principal est autosomique récessif. On évoque plusieurs hypothèses pour expliquer le développement précoce des dilatations kystiques des voies biliaires qui la caractérisent : une occlusion néonatale de l’artère hépatique à l’origine d’ischémie biliaire, sténose et dilatations secondaires (hypothèse peu probable qui explique mal les dilatations fusiformes visibles sans sténose), un développement anormal de l’épithélium biliaire et du tissu avoisinant, et une involution incomplète de la plaque biliaire qui entoure les branches portales au cours du développement embryonnaire (4). La maladie de Caroli est associée à un risque accru de cholangiocarcinome, 100 fois supérieur à celui de la population générale, dont l’incidence est de 5 % à 10 %. Bien que présente dès la naissance, la maladie peut rester longtemps asymptomatique, pendant les 20 premières années, voire tout au long de la vie. Elle peut au contraire se manifester dès l’enfance. Lorsqu’ils sont symptomatiques, la majorité des patients ont une franche altération de leur qualité de vie. La maladie se manifeste fréquemment par une fièvre récidivante, un ictère et/ ou des douleurs de l’hypochondre droit. L’angiocholite aiguë est en effet le principal mode de présentation (64 % des patients) et la première cause de mortalité de ces patients. Habituellement causée par des bacilles à Gram négatif, elle récidive fréquemment et finit par résister aux antibiotiques. L’hypertension portale secondaire à la fibrose hépatique congénitale est présente dans 20 % à 50 % des cas et peut se compliquer d’hypersplénisme et de rupture de varices œsophagiennes (20).
Biologiquement, on observe généralement une cholestase chronique. La fonction hépatique reste longtemps préservée. L’hyperleucocytose à polynucléaires neutrophiles, possible malgré l’hypersplénisme, s’observe au cours des épisodes d’angiocholite.
Imagerie
La clé du diagnostic radiologique de la maladie de Caroli repose sur la démonstration d’une communication entre les lésions kystiques et les canaux biliaires normaux (6) (figure 3A). Cela peut être vu en échographie ou en tomodensitométrie, mais c’est la cholangio-IRM qui est la plus rentable, car elle visualise l’intégralité des voies biliaires. Trois aspects principaux de maladie de Caroli sont décrits en cholangio-IRM : 1) une forme associant multiples ectasies kystiques et dilatations fusiformes (figure 3B) ; 2) une forme associant dilatations fusiformes isolées avec calculs multiples ; et 3) une forme localisée au lobe gauche, associant dilatations, kystes et calculs multiples (4). Au scanner avec injection, outre la communication des kystes avec voies bilaires, on décrit un signe spécifique : le dot sign. Il correspond à une branche portale de petite taille entourée de voies biliaires dilatées (6). Ce signe peut également être mis en évidence en IRM (figure 3C) ou en écho-doppler. La CPRE à visée diagnostique est contre indiquée car elle peut favoriser les épisodes d’angiocholite. Dans les formes associées à la fibrose hépatique congénitale, le foie est dysmorphique et les signes morphologiques en rapport avec l’hypertension portale, au premier rang desquels les voies de dérivation veineuses, peuvent être au premier plan. L’ensemble de ces anomalies rend très difficile le diagnostic de cholangiocarcinome, dont le risque d’apparition justifie un suivi morphologique régulier des patients atteints de maladie de Caroli. La comparaison des cholangio-IRM successives peut ainsi permettre de mettre en évidence un syndrome de masse ou une distorsion des voies biliaires dilatées.
Traitement
Le traitement de la maladie de Caroli dépend de ses conséquences cliniques et de la localisation de l’atteinte biliaire. Le traitement conservateur doit être privilégié, associant antibiothérapie lors des épisodes d’angiocholite, acide ursodésoxycholique, et traitement endoscopique pour extraction des calculs intrahépatiques accessibles. La chirurgie est réservée aux formes symptomatiques ayant résisté aux traitements conservateurs. Dans les formes localisées au foie droit ou, le plus souvent, au foie gauche, une hépatectomie droite ou gauche sera proposée. La morbidité est faible, la mortalité quasiment nulle, et la survenue de tumeurs malignes après résection hépatique n’a jamais été décrite (4). Lorsque l’atteinte est diffuse sur les deux lobes, qu’il existe des angiocholites à répétition, des abcès, des septicémies, a fortiori associées à une cirrhose biliaire secondaire, l’hépatectomie n’est plus possible et la transplantation hépatique doit être proposée (21). Elle peut être réalisée malgré la présence d’une infection biliaire tant que le patient est stable. Ses résultats sont satisfaisants à long terme, avec une survie à 5 ans comprise entre 77 et 86 % dans les 3 séries publiées. La transplantation a aussi le mérite de prévenir le développement du cholangiocarcinome, et la maladie de Caroli ne récidive jamais sur le greffon. Lorsque qu’un cholangiocarcinome est présent, la transplantation est contre-indiquée compte tenu d’un taux de récidive élevé post transplantation. Lorsqu’une fibrose hépatique congénitale est présente, la transplantation peut aussi être le recours aux formes d’hypertension portale invalidantes, avec ascite réfractaire ou hémorragies digestives récidivantes non contrôlées par la mise en place d’un TIPS, ou contrôlées au prix d’une encéphalopathie hépatique chronique. Dans tous les cas, un accès dérogatoire au greffon doit être demandé à l’agence de la Biomédecine sous la forme d’une « composante expert » (21).
Polykystose hépatique
La polykystose hépatique (PKH) est une maladie héréditaire rare et caractérisée par la présence de multiples kystes diffus du foie. Elle peut être isolée (PKH isolée) ou, le plus souvent, associée à la polykystose rénale autosomique dominante (PKRAD), ou à la polykystose rénale autosomique récessive (PKRAR), beaucoup plus rare. Les mécanismes impliqués dans la genèse des kystes ne sont pas encore tous parfaitement élucidés. On sait que dans la PKRAD, ils impliquent un dysfonctionnement des cils primaires des cellules épithéliales biliaires, qui régulent la concentration de calcium intracellulaire. La baisse de calcium intracellulaire entraîne l’activation de plusieurs voies qui stimulent la prolifération des cholangiocytes et la sécrétion du fluide kystique. Les polykystoses sont ainsi classées dans le cadre nosologique des ciliopathies (22). La dysplasie congénitale des voies biliaires observée dans la PKH pourrait toutefois avoir d’autres mécanismes non encore élucidés, raison pour laquelle la PKH est encore souvent classée simplement parmi les maladies kystiques congénitales des voies biliaires intrahépatiques, au même titre que les hamartomes ou la maladie de Caroli.
Épidémiologie et génétique
PKH associée à la polykystose rénale autosomique dominante (PKRAD)
La PKRAD est une maladie génétique monogénique fréquente, dont la prévalence est de l’ordre de 1/1 000. La PKH est le symptôme extrarénal le plus courant de la PKRAD, qui concerne 90 % des patients atteints de PKRAD. Des mutations dans deux gènes (PKD1 et PKD2) provoquent le développement de la PKRAD. Le gène PKD1 est situé sur le chromosome 16 et code pour une protéine appelée polycystine-1 (PC1). Des mutations de ce gène sont retrouvées chez plus de 80 % des malades atteints de PKRAD. Le gène PKD2 est situé sur le chromosome 4, et est responsable des cas restants. Il code pour la polycystine-2 (PC2). PC1 et PC2 sont exprimées dans le cil primaire des cellules tubulaires rénales et de cholangiocytes. PC1 agit comme un mécanorécepteur sensible aux flux liquidiens. PC2 est complexée à PC1 et le complexe PC1-PC2 régule la concentration de calcium intracellulaire. PC2 est aussi impliquée dans la maturation glycoprotéique de PC1 dans le réticulum endoplasmique.
En cas de PKRAD, il existe une défaillance dans la production, la maturation et l’adressage de PC1 aboutissant à la baisse de la concentration de calcium intracellulaire. Cette baisse de concentration calcique active les voies mTOR, MAP-kinase, WNT et β-caténine qui stimulent la prolifération cellulaire et la sécrétion du fluide kystique. D’autres mutations ont été décrites, touchant les gènes LRP5 (dont la protéine interagit avec les protéines transmembranaires Frizzled pour moduler la transduction de la voie WNT/β-catenine), GANAB (impliqué dans le repliement des protéines), ALG9 (impliqué dans la glycosylation des protéines), et DZIP1L (impliqué dans le transport de PC1 et PC2 à la membrane du cil primaire) (23).
PKH associée à la polykystose rénale autosomique récessive (PKRAR)
La PKRAR est assez rare avec une incidence d’environ 1/20 000. Elle survient souvent chez les enfants, dont 30 % meurent d’une dysplasie pulmonaire sévère et d’une insuffisance respiratoire secondaire, associée à une dilatation du canal collecteur rénal, une dysplasie des voies biliaires et une fibrose portale. À l’heure actuelle, une mutation du gène PKHD1 sur le bras court du chromosome 6 codant pour la fibrocystine (FC), une protéine dont la fonction est encore mal connue, mais qui pourrait être impliquée la stimulation de PC2, s’avère être la principale anomalie génétique associée à la PKRAR (24).
PKH isolée ou PKHAD
La PKH isolée est une maladie à transmission autosomique dominante à pénétrance faible (<5 %). Les mutations concerneraient principalement le gène PRKCSH (15 %), suivie par SEC63 (10 %), ALG8 (2 %) PKHD1, LRP5, SEC61B et GANAB (environ 1 %). PRKCSH code pour une protéine du complexe de la glucosidase II, la glucosidase IIβ (GluIIβ), impliquée dans la glycosylation protéique. SEC63 code pour une protéine impliquée dans la conformation, le transport et la dégradation protéique au sein du réticulum endoplasmique. Le gène ALG8 code pour une protéine membranaire du réticulum endoplasmique, et le produit codé par le gène SEC61B est un composant important du complexe protéique SEC63 dans le réticulum endoplasmique. Les deux gènes jouent un rôle important dans la régulation de la qualité des protéines. Dans un grand nombre de cas, le gène pathogène ne peut pas être trouvé (25). Il pourrait concerner d’autres phénomènes que ceux liés à la maturation et l’adressage des protéines du cil primaire (24). Récemment, il a ainsi été montré que l’autophagie des cholangiocytes contribuait à la cystogenèse hépatique dans la PLD isolée et pourrait ainsi représenter une cible thérapeutique potentielle (25).
Place du diagnostic génétique en clinique
Au cours de la polykystose rénale autosomique dominante, le diagnostic génétique peut être utile dans les formes débutantes ou ambiguës ne répondant pas aux critères diagnostiques de Ravine, qui ont été élaborés à partir des caractéristiques des PKRAD de type PKD1 (26). Le diagnostic génétique peut alors confirmer une PKRAD de type PKD2, de meilleur pronostic compte tenu de son début plus tardif et de l’apparition d’une insuffisance rénale terminale en moyenne 20 ans après comparé aux formes PKD1, confirmer, chez l’enfant, une forme PKD1 débutante, ou réorienter le diagnostic (27). Dans le cas de formes avec quelques kystes rénaux et une atteinte polykystique hépatique, le diagnostic génétique, en affirmant l’absence de mutation PKD1 et PKD2, et la présence d’un variant PRKCSH, peut confirmer le diagnostic de polykystose hépatique isolée et affirmer un risque nul d’insuffisance rénale (27). Néanmoins, seules 30 à 45 % des PKH isolées ont une mutation connue, et c’est la principale raison pour laquelle il n’existe pas actuellement de recommandation pour prescrire un diagnostic génétique en cas de PKH.
Manifestations Cliniques
Au cours de la polykystose hépatique ou hépato-rénale, les kystes hépatiques sont rares avant l’âge de 20 ans et présents dans plus de 80 % des cas après 60 ans. Le volume hépatique augmente d’environ 2 % tous les 6 à 12 mois, mais la majorité des patients restent asymptomatiques, quel que soit le type de PKH (24). Seuls 20 % des patients vont développer des symptômes : dyspnée due au refoulement thoracique ou à la compression des cavités cardiaques droites, œdèmes par compression de la veine cave inférieure, satiété précoce, distension abdominale, reflux gastro-œsophagien, dénutrition avec amaigrissement et sarcopénie secondaires à la compression gastrique chronique, douleurs dorsales ou de l’hypochondre droit dues à une hépatomégalie pressant les organes environnants ou à des complications kystiques, ou douleurs électives secondaires à des kystes de localisation mal tolérée (sous le rebord costal ou à l’épigastre), qui affecteront gravement la qualité de vie (21,24). À ces manifestations dues à l’effet de masse kystique s’ajoute une altération du parenchyme hépatique non kystique due à une obstruction de l’écoulement veineux sus-hépatique, entraînant une hypertension portale qui peut se compliquer d’ascite, d’hémorragie variqueuse ou d’hypersplénisme. Une série rétrospective française ayant examiné les pièces de résection hépatique ou les explants a quantifié à 92 % la fréquence de la dilatation sinusoïdale au sein du parenchyme hépatique « sain », témoignant de l’obstacle au retour veineux (28). Les kystes hépatiques n’entrainent jamais d’insuffisance hépatique. On observe fréquemment des anomalies biologiques hépatiques, principalement d’allure cholestatique, en rapport avec la compression des canaux biliaires, l’activation des cellules biliaires, ou les anomalies de la microcirculation. 45 % des patients atteints de polykystose hépatique ont une augmentation du CA19-9 avec un degré d’élévation positivement corrélé au volume du foie polykystique (29).
Les facteurs de risque de la survenue de symptômes chez les patients atteints de PKRAD sont l’âge avancé (effet plus marqué pour les hommes que pour les femmes), et surtout le sexe féminin. En moyenne, les femmes sont de 9 ans plus jeunes que les hommes lors du diagnostic de la PKH, et 91 % des femmes seront symptomatiques contre seulement la moitié des hommes. Chez la femme ménopausée non substituée, l’expansion du volume hépatique diminue alors que les kystes hépatiques se développent rapidement sous traitement hormonal substitutif (30). L’effet de la contraception œstroprogestative sur l’expansion des kystes hépatiques est moins spectaculaire probablement du fait des faibles doses d’estrogènes administrées et des formes moins évoluées de polykystoses chez les jeunes femmes concernées. Il serait toutefois significatif et justifie de déconseiller la contraception orale chez les femmes atteintes de polykystose hépatique. L’influence des œstrogènes sur l’histoire naturelle des polykystoses peut être expliquée par la présence des récepteurs aux œstrogènes α et β à la surface des cellules épithéliales kystiques, contrairement aux cellules épithéliales des canaux biliaires normaux. Chez les souris, ces récepteurs sont surexprimés en cas de ligature du cholédoque et impliqués dans la prolifération et la sécrétion cholangiolaire par un mécanisme impliquant l’insulin growth factor 1 (IGF-1). Au cours des polykystoses, il existe une expression d’IGF1 détectable histologiquement à la surface des kystes, qui suggère qu’un phénomène identique se produit chez les femmes atteintes de PKH (31).
La polykystose hépatique peut se compliquer d’accidents aigus hémorragiques ou infectieux. L’hémorragie intrakystique est fréquente et entraîne des douleurs dont la durée est corrélée à la taille des kystes hémorragiques concernés. Elle peut récidiver et entraîner une anémie ferriprive (32). L’infection des kystes hépatiques, (dont l’incidence est de 0,1 %/patient/an) concerne le plus souvent une entérobactérie. Elle entraine une fièvre, un syndrome inflammatoire biologique, et une augmentation significative de CA19-9, dont la diminution témoigne de l’efficacité du traitement antibiotique (24).
Imagerie
Le diagnostic de PKH est généralement posé lorsque le nombre de kystes hépatiques est supérieur à 20, ou supérieur à 4 chez un patient aux antécédents familiaux de PKHAD isolée (24). Le type de PKH peut être difficile à identifier car les patients atteints de PKHAD peuvent avoir des kystes rénaux, tandis que les patients atteints de PKRAD ou de PKRAR peuvent avoir des kystes hépatiques seuls en cause dans l’expression clinique de la maladie polykystique.
En imagerie, les kystes, de taille variable mais souvent supracentimétriques, ont toutes les caractéristiques des kystes simples : bien limités, à parois fines, sans cloison ni végétation murale, et avec un renforcement postérieur à l’échographie. Lorsque le nombre de kystes est très important, l’architecture du foie est remaniée, entraînant une perte des repères anatomiques et vasculaires habituels (1). Les remaniements intrakystiques secondaires aux hémorragies et aux infections sont fréquents. En imagerie en coupe, la paroi des kystes ne se rehausse pas après injection de produit de contraste et on observe rarement des calcifications (figure 4). L’IRM est surtout utile en cas de complication, pour visualiser un signal hyperintense en T1, signe d’hémorragie intrakystique (1). La localisation de kystes infectés peut être difficile en imagerie conventionnelle du fait de la fréquence élevée de remaniements kystiques secondaires à des épisodes hémorragiques antérieurs. Le PET-scan peut alors être très utile pour localiser le ou les kystes infectés, ou infirmer le diagnostic d’infection kystique en identifiant un foyer infectieux extrahépatique (23).
L’imagerie permet de dénombrer les kystes, préciser leur taille et le volume de parenchyme épargné par les kystes. Ces paramètres sont inclus dans les classifications de Gigot (1997) et de Schnelldorfer (2009) (Tableau 2).
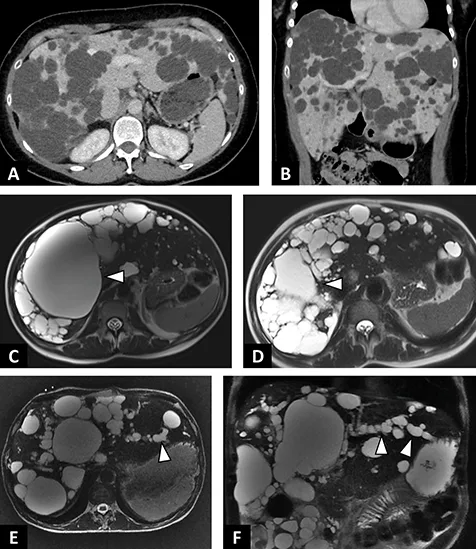
Figure 4 : Exemples de polykystoses hépatiques |
| A–B : Polykystose hépatique non compliquée. A. Scanner abdominal en coupe axiale après injection de produit de contraste au temps portal. Multiples kystes hépatiques hypodenses, bien limités intéressant l’ensemble des segments hépatiques. B. Même scanner, reconstruc tion coronale. Augmentation en volume du foie conséquences des multiples kystes biliaires. C–D : Polykystose hépatique chez une patiente symptomatique de 44 ans. C. IRM en séquence axiale T2 sans saturation du signal de la graisse. Kyste prédominant du foie droit (tête de flèche). D. IRM en séquence axiale T2. Contrôle 3 mois après alcoolisation percutanée du kyste prédominant. E–F : Polykystose hépatique chez un patient de 54 ans révélée par un ictère non fébrile. E. IRM en séquence axiale T2 avec saturation du signal de la graisse. Polykystose hépatique avec multiples kystes périhilaires comprimant le hile hépatique associé à une dilatation des voies biliaires d’amont (tête de flèche). F. IRM en séquence coronale T2 avec saturation du signal de la graisse. Dilatation des voies biliaires intra hépatiques du lobe gauche (tête de flèche). |
Traitement
Traitement médical
Les deux seuls traitements médicaux ayant montré un intérêt au cours des polykystoses hépatiques sont les analogues de la somatostatine et l’acide ursodésoxycholique. Les inhibiteurs de mTOR ont été étudiés dans cette indication après qu’une réduction du volume hépatique a été rapportée chez certains greffés rénaux atteints de polykystose hépatique traités par sirolimus mais les essais thérapeutiques évaluant les inhibiteurs de mTOR seuls ou en association avec un analogue de la somatostatine n’ont finalement pas été concluants (24).
Analogues de la somatostatine
Les analogues de la somatostatine représentent une stratégie thérapeutique potentielle en raison de leur capacité à supprimer la production d’AMPc, et par conséquent la sécrétion du liquide intrakystique. Plusieurs études ont rapporté les effets bénéfiques attribués à l’administration d’analogues de la somatostatine à la fois dans des modèles animaux de PKH et chez des patients. Chez des rats traités à l’octréotide, une réduction significative des taux d’AMPc, du volume kystique hépatique, du poids du foie, et de la fibrose sont observés corrélés à la dose et à la durée du traitement (33). Plusieurs essais cliniques ont été réalisés pour évaluer l’efficacité et l’innocuité de l’octréotide et du lanréotide chez les malades atteints de PKH associée ou non à une PKR. L’effet maximal est observé pendant les 6 premiers mois sous la forme d’une réduction significative mais modérée (-5 %) du volume total du foie, associée à une amélioration de la qualité de vie. Ces bénéfices seraient surtout observés chez les femmes. Malheureusement, plusieurs effets secondaires (calculs vésiculaires, diarrhée, nausées, vomissements, étourdissements et maux de tête) limitent l’utilisation de ces molécules, tout comme l’absence d’effet durée : un traitement prolongé de 12 mois pour le lanréotide ou jusqu’à 4 ans pour l’octréotide, n’a en effet pas donné de meilleurs résultats qu’un traitement de 6 mois. Le seul intérêt du traitement prolongé serait de maintenir les effets bénéfiques et prévenir un effet rebond sur le volume hépatique (22). Malheureusement, les patients traités plus de 18 mois par des analogues de la somatostatine peuvent perdre du poids et développer une sarcopénie (34). En conclusion, malgré leur rationnel séduisant, les analogues de la somatostatine ont un intérêt limité en pratique.
Tableau 2 : Classifications des polykystoses hépatiques| Classificatrion de Gigot |
|---|
| Nombre de kystes | taille | Volume de Parenchyme épargné |
Gigot type I
Gigot type II
Gigot type III | < 10
Multiples
Multiples | > 10 cm
Petite ou moyenne
Petite ou moyenne | Important
Important
Faible |
| Classification de Schnelldorfer des polykystoses hépatiques | |
|---|
| Symptomes | Caractéristiques des kystes | Volume
de Parenchyme épargné | Obstruction portale ou sushépatique dans le territoire préservé par les kystes |
Type A
Type B
Type C
Type D | Absents ou discrets
Modérés à sévères
Sévères
Sévères | Indifférent
Nombre limité, Gros kystes
Indifférent
Indifférent | Indifférent
> 2 secteurs
> 1 secteur
< 1 secteur | Indifférent
Absente
Absente
Présente |
Acide ursodésoxycholique
L’acide ursodésoxycholique (AUDC) est capable d’inhiber l’hyperplasie biliaire caractéristique des modèles expérimentaux de cholestase obstructive. Au cours des PKH, l’effet de l’AUDC a été évalué dans des modèles précliniques et dans un seul essai clinique. L’administration chronique d’AUDC à de jeunes rats atteints de PKH a bloqué la cystogenèse et la fibrose hépatique et a amélioré leur condition physique. Un effet inhibiteur de l’AUDC sur l’hyperprolifération des cholangiocytes kystiques était observé tant à l’état basal qu’après stimulation par des acides biliaires promitotiques. L’AUDC a diminué la concentration hépatique des acides biliaires les plus cytotoxiques et a normalisé la concentration des acides biliaires dans la bile des rats atteints de PKH. Ces effets bénéfiques ont été associés à la normalisation du calcium intracellulaire dans les cholangiocytes kystiques (35). Ces données positives ont encouragé la réalisation d’un essai clinique international multicentrique de phase 2 pour évaluer l’effet de l’AUDC (15 à 20 mg/kg/j pendant 6 mois) chez les patients atteints de PKH symptomatiques avec une maladie avancée (> 20 kystes et volume hépatique > 2 500 ml). Globalement, l’AUDC a entraîné une diminution de la GGT et des phosphatases alcalines, mais n’a pas entrainé de modification significative du volume hépatique. En analyse de sous-groupes, une augmentation significative à la fois du volume hépatique (+4,6 %) et du volume des kystes hépatiques (+20 ml) était observée chez les patients atteints de PKRAD non traités, alors qu’aucune modification de ces paramètres n’était constatée sous AUDC. Cet effet bénéfique observé chez les patients atteints de PKRAD n’était pas constaté chez les patients atteints de PKH isolée (36).
Traitement percutané
La ponction aspiration et sclérose telle que décrite plus haut pour les kystes biliaires symptomatiques peut aussi être réalisée en cas de douleurs focales chroniques en rapport avec un kyste de plus de 4 cm mal situé (figures 4C,4D). Elle est efficace dans 66 % des cas mais des récidives tardives peuvent s’observer, justifiant de répéter le geste. Le drainage des kystes infectés est indiqué an cas d’échec du traitement antibiotique.
L’embolisation artérielle, destinée à interrompre la vascularisation des kystes (qui provient exclusivement de l’artère hépatique), a montré dans quelques études asiatiques une réduction du volume des kystes comprise entre 33 % et 90 % à 1 an, avec un effet positif sur les symptômes. Ces résultats n’ont néanmoins pas été reproduits par d’autres auteurs, qui ont rapporté un taux d’échec de 70 % et un risque d’insuffisance hépatique. L’embolisation artérielle n’a donc pas encore de place dans l’arsenal thérapeutique des polykystoses hépatiques. Elle pourrait à l’avenir faire l’objet d’essais contrôlés versus chirurgie ou être testée comme traitement adjuvant (24).
Traitement chirurgical
Fenestration
Contrairement à la ponction aspiration sclérose, qui s’adresse à seul kyste qui pose problème, la fenestration est souvent utilisée pour le traitement de plusieurs kystes, c’est-à-dire pour des patients classés Gigot I-II ou Schnelldorfer B. La fenestration est généralement réalisée par voie laparoscopique, mais elle doit parfois être réalisée par chirurgie à ciel ouvert en raison d’un saignement incontrôlable, d’angles morts ou d’impératifs techniques (37). Les complications courantes de cette procédure comprennent l’ascite post-opératoire, l’épanchement pleural, l’hémorragie et les fuites de bile. Les symptômes sont grandement soulagés dans 90 % des cas de fenestration, mais 33,7 % des patients souffrent de récidive symptomatique et 26,4 % ont besoin d’une nouvelle intervention. Les patients ayant plusieurs kystes de plus de 5 cm de diamètre ont un taux de récidive plus élevé que les patients ayant des kystes de plus petit volume.
Résection hépatique
L’hépatectomie est souvent utilisée chez les patients atteints de PKH Gigot II sévère avec au moins un segment du foie qui n’est épargné par les kystes. L’étendue de la résection dépend de la taille et de la distribution des kystes. La déformation de la capsule de Glisson et l’obstruction veineuse hépatique augmentent la difficulté de la résection. C’est la raison pour laquelle la fenestration de kystes inextirpables accompagne souvent la résection hépatique (24). L’obstruction veineuse sus-hépatique fréquente dans la PKH augmente les saignements peropératoires et influence de façon majeure la morbidité post-opératoire en favorisant l’ascite et l’insuffisance hépatique (28). Dans l’étude rétrospective de Chebib et al. incluant 186 patients PLD ayant subi une hépatectomie, le volume hépatique postopératoire était réduit en moyenne de 61 % par rapport au volume hépatique préopératoire, au prix d’un taux de complications élevé de 21 % et un taux de mortalité de 2,7 % (38). Certaines études affirment néanmoins que l’hépatectomie peut atténuer grandement les symptômes et limiter ou retarder le recours à la transplantation hépatique même si celle-ci pourrait être de réalisation plus difficile en raison des adhérences sus mésocoliques induites par la résection hépatique. L’administration d’analogues de la somatostatine après l’hépatectomie pourrait être utile, en inhibant la croissance des kystes résiduels et en prévenant l’apparition de nouveaux kystes. Malgré tous ces arguments, chez les patients candidats, la balance bénéfice-risque penche actuellement plutôt pour une transplantation hépatique qu’une résection hépatique, cette dernière étant réservée à un petit nombre de patients sélectionnés dans des centres experts habitués à la technique et aux suites opératoires difficiles (21).
Transplantation hépatique
La transplantation hépatique est actuellement le seul traitement curatif de la PKH. Elle concerne principalement les patients classés Gigot III ou Schnelldorfer D, présentant des symptômes sévères qui affectent la qualité de vie, ainsi que des complications non traitées telles les hémorragies intrakystiques récidivantes responsables d’anémie sévère, les infections kystiques à germes multirésistants, l’ascite réfractaire compliquant l’hypertension portale liée au bloc sus hépatique, et surtout la dénutrition et la sarcopénie (21). Il ne faut en aucun cas attendre la survenue d’une insuffisance hépatique pour poser l’indication de la transplantation car la fonction hépatique est préservée au cours de la polykystose hépatique. Malgré le contexte de dénutrition et d’infection possible des kystes lors de la greffe, les résultats à long-terme rapportés dans les deux études publiées à ce jour sont très satisfaisants, supérieurs à ceux de la transplantation pour cirrhose ou carcinome hépatocelllulaire (39,40) : le taux de survie à 5 ans était en effet de 92,3 % dans la première étude, et de 85,1 % dans la seconde. La majorité des patients récupèrent une bonne qualité de vie après la transplantation (41) et la polykystose ne récidive jamais sur le greffon.
En cas de polykystose hépatique et rénale, la transplantation combinée foie-rein doit être d’emblée envisagée dès qu’il existe une indication de transplantation rénale et une volumineuse polykystose hépatique, a fortiori symptomatique. La double greffe peut être envisagée avant le recours à la dialyse. L’utilisation du rein et du foie provenant du même donneur s’accompagne en effet d’un bénéfice immunologique qui diminue le risque de rejet rénal. La survie à 5 ans après transplantation combinée est excellente, évaluée à 90 % d’après les données du registre américain (42). En France, le système dérogatoire de la composante expert permet un accès au greffon hépatique dans les 6 mois qui suivent l’inscription sur la liste d’attente (21).
Tumeurs mucineuses kystiques
Ces lésions rares étaient auparavant connues sous les noms de cystadénome et de cystadénocarcinome. Elles ont été reclassées en deux entités différentes par la classification OMS 4e et 5e édition des tumeurs de l’appareil digestif en 2010 : 1) les tumeurs mucineuses kystiques (TMK) du foie et des voies biliaires lorsqu’un stroma de type ovarien est présent et que la lésion ne communique pas avec l’arbre biliaire ou 2) les tumeurs papillaires intracanalaires (TPI) du foie et des voies biliaires lorsqu’il existe une communication avec les voies biliaires. La nouvelle classification de l’OMS imposant la présence d’un stroma de type ovarien pour parler de tumeur mucineuse kystique, certaines tumeurs kystiques qui n’en possèdent pas, en particulier chez les hommes, auparavant classées parmi les cystadénomes, sont reclassées parmi les tumeurs papillaires intracanalaires (43).
Épidémiologie, origine et présentation clinique
Les tumeurs mucineuses kystiques sont rares et représentent moins de 5 % de l’ensemble des lésions kystiques intrahépatiques, moins de 5 % des tumeurs biliaires, et moins de 0,5 % des tumeurs malignes du foie (4). On estime leur prévalence comprise entre 1/10 000 et 1/100 000 (1). La présence de stroma ovarien dans les TMK et son aspect morphologique similaire à celui des tumeurs mucineuses kystiques ovariennes suggèrent que les TMK proviennent d’un stroma mésenchymateux ovarien ectopique qui migre vers le foie ou le pancréas au cours du développement embryonnaire. Il a été observé que l’épithélium de surface péritonéale des gonades embryonnaires est tapissé de cellules bombées par opposition à l’épithélium cœlomique aplati des autres organes de la partie supérieure de l’abdomen. Cette morphologie distincte suggère que ces cellules bombées ont la capacité de se détacher de la surface gonadique et de se fixer à la surface péritonéale des organes voisins au cours de la cinquième à la huitième semaine de développement, lorsque les gonades embryologiques sont situées juste en dessous du diaphragme et à proximité du foie, de la rate et de la queue du pancréas (44). Une autre hypothèse est que l’épithélium dérivé de l’endoderme et du mésenchyme primitif dans le pancréas et le foie fœtaux deviennent hypersensibles aux stéroïdes sexuels féminins et sont amenés à proliférer.
La plupart des TMK surviennent sans carcinome invasif associé, le plus souvent chez les femmes caucasiennes d’âge moyen, (sex ratio> 9/1). Cependant, le risque de carcinome invasif associé aux TMK serait plus élevé chez les hommes que chez les femmes. Ces lésions peuvent provoquer des douleurs, un effet de masse (principalement une compression gastrique), une obstruction des voies biliaires, ou être asymptomatiques et découvertes fortuitement. La présence d’un stroma de type ovarien dans le TMK (ou dans la plupart des cystadénomes si on s’en réfère à l’ancienne terminologie) explique probablement la forte prépondérance de ces lésions chez les femmes. La raison pour laquelle certains hommes peuvent développer une TMK n’est pas claire. On invoque une similitude entre le stroma caractéristique de type ovarien et les cellules stromales observées dans le tractus hépatobiliaire embryonnaire (43).
Figure 5 : Exemples de tumeurs mucineuses kystiques |
A–B : Tumeur mucineuse kystique de bas grade (anciennement cystadénome mucineux) chez une patiente de 37 ans. A. Scanner abdo minal en coupe axiale, temps portal. Lésion kystique arrondie, exophytique du segment V, bien limitée, contenant de fines cloisons rehaussées après injection de produit de contraste (tête de flèche). B. Même examen, reconstruction coronale. Cloisons intrakystiques rehaussées après injection (tête de flèche).
C : Tumeur mucineuse kystique de haut grade (anciennement cystadénocarcinome). Scanner abdominal au temps portal en coupe axiale. Volumineuse lésion kystique du segment IV contenant des végétations intrakystiques irrégulières et rehaussées après injection de produit de contraste (tête de flèche).
D–F : Tumeur mucineuse kystique chez une patiente de 57 ans. Aspects scanner et IRM. D. Scanner abdominal en coupe axiale après injec tion de produit de contraste au temps portal. Volumineuse lésion kystique du foie droit (flèche) refoulant le reste du foie à gauche. E. Même examen, reconstructions coronales. Cloisons intrakystiques rehaussées après injection de produit de contraste à la partie inférieure de la lésion, associées à de fines calcifications des cloisons (cercle rouge). F. IRM en séquence axiale T2 avec saturation du signal de la graisse. Cloisons en hyposignal T2 (cercle rouge) au sein d’une lésion kystique en hypersignal T2 liquidien. |
Imagerie
Les TMK sont généralement de grande taille. À l’échographie conventionnelle, elles apparaissent généralement hypoéchogènes, avec des parois irrégulières épaissies et des échos internes occasionnels. Elles peuvent être décrites comme une masse kystique uniloculaire bien définie, mais la description typique est celle d’une masse multiloculaire, avec des nodules muraux et des septa (6). En échographie de contraste, on observe un rehaussement septal au temps artériel qui disparaît aux temps portal et tardif (4). Le cystadénocarcinome a des caractéristiques identiques, avec une présence supposée constante de septa et de nodules septaux et muraux, prenant le contraste au temps artériel et d’épaisseur pouvant dépasser 1 cm (45). Des calcifications épaisses et grossières peuvent aussi être décrites sur les septa en échographie conventionnelle. Il est en pratique très difficile de distinguer une TMK bénigne d’un cystadénome. Les caractéristiques de réhaussement sont identiques. L’absence de nodule mural et l’aspect uniloculaire seraient les deux caractéristiques qui, si elles sont présentes, discriminent le mieux une TMK bénigne d’un cystadénocarcinome.
Il se pose alors la difficulté de distinguer ces TMK de kystes biliaires simples.
Au scanner (figures 5A, 5B, 5C, 5D, 5E), les caractéristiques sont les mêmes à la différence près que la présence de cloisons pourrait être plus souvent décrite qu’en échographie, jugée constante dans une expérience monocentrique américaine rassemblant 22 TMK diagnostiquées sur 17 ans (46). La paroi du kyste est généralement épaissie ou irrégulière, contrairement à celle d’un kyste simple, mais cette caractéristique peut manquer. Une dilatation périkystique des voies biliaires intrahépatiques est possible.
L’imagerie par résonance magnétique (IRM) met en évidence une lésion bien définie hypointense en T1, hyperintense en T2 (signal liquidien), qui ne se rehausse pas après l’administration de gadolinium par voie intraveineuse. L’architecture interne (nodules et cloisons) est bien identifiée (figure 5F). Un hypersignal spontané en T1, évoquant la présence d’un liquide hémorragique intralésionnel, est parfois considéré comme un signe de malignité (1). Il n’existe pas de communication avec les voies biliaires. L’IRM ne permet pas de mettre en évidence spécifiquement le stroma pseudo ovarien.
Anatomopathologie
Macroscopiquement, les TMK ont le plus souvent l’apparence d’un kyste multiloculaire solitaire cloisonné. Seuls 10 % se présentent sous la forme d’un kyste uniloculaire. Ils sont le plus souvent supracentimétriques, volumineux, mesurant jusqu’à 30 cm de diamètre. Le liquide du kyste est mucineux et teinté de bile. La surface interne peut être lisse ou trabéculée, et rarement, le siège de nodules muraux papillaires. Les tumeurs ne sont pas connectées à l’arbre biliaire (44).
Microscopiquement, la surface interne du kyste est composée d’un épithélium monocouche de cellules cubiques à cylindriques, produisant de la mucine, ressemblant à l’épithélium biliaire. Un stroma de type ovarien composé de cellules fusiformes est présent sous l’épithélium. Ce stroma exprime les récepteurs aux œstrogènes, à la progestérone et à l’alpha-inhibine, et peut présenter des cellules épithélioïdes ressemblant à des cellules lutéinisées. Les cellules épithéliales expriment le plus souvent un immunophénotype de type biliaire, (CK7, CK8, CK18, CK19, et ACE), et accessoirement une différenciation gastro-intestinale (47). L’expression de marqueurs gastro-intestinaux, tels que CK20, MUC2, MUC5AC et MUC6, a été associée à un grade croissant de dysplasie et de carcinome invasif (44). La majorité des TMK présentent une dysplasie de bas grade. Les TMK avec un carcinome invasif associé sont rares, représentant environ 6 à 8 % des cas. Comme ce qui est observé dans le pancréas, les tumeurs dégénérées sont plus grosses, avec des projections papillaires grossières.
Figure 6 : Diverses lésions kystiques hépatiques « pièges » et fréquentes |
A–B : Métastases kystiques d’une tumeur neuroendocrine rectale découvertes chez un patient de 54 ans à l’occasion de douleurs de l’hypochondre droit. A. Scanner abdominal en coupe axiale après injection de produit de contraste au temps portal. Multiples lésions kystiques hépatiques avec végétations intrakystiques (cercle rouge) en rapport avec lésions secondaires. B. Même examen, coupes inférieures. Multiples lésions secondaires kystiques hépatiques avec végétations intrakystiques (cercle rouge).
C : Métastase kystique d’une GIST gastrique. Scanner abdominal en coupe axiale après injection de produit de contraste au temps portal. Lésion mixte, tissulaire et kystique du foie droit avec paroi hypervasculaire (tête de flèche).
D–E : Kyste biliaire multiloculé mimant un cystadénome mucineux chez une patiente de 67 ans. D. IRM en séquence axiale T2 avec saturation du signal de la graisse. Lésion kystique polylobée, contenant des cloisons intrakystiques en hyposignal T2 et une indentation en regard des cloisons (flèches). E. IRM en séquence axiale T1 après injection de gadolinium au temps portal montrant un fin rehaussement des cloisons intrakystiques (cercle rouge).
F : Kystes péri-biliaires chez un patient atteint de cirrhose alcoolique IRM en séquence axiale T2. Multiples lésions kystiques centimétriques accolés aux voies biliaires proximales (tête de flèche). |
Diagnostics différentiels
Le diagnostic différentiel préopératoire entre les TMK et les autres lésions peut être très difficile en imagerie tant il existe des situations pièges. Les trois principales lésions pouvant faire évoquer à tort une TMK sont les abcès hépatiques, les kystes hydatiques, et surtout kystes biliaires simples d’aspect atypique. Le contexte septique et l’exposition endémique sont des éléments clés d’orientation diagnostique pour les deux premières lésions. La sérologie hydatidose confirmera le diagnostic de kyste hydatique. Pour le diagnostic bactériologique de l’abcès, la ponction exploratrice est indispensable, alors qu’elle est contre indiquée en cas de kyste hydatique car une fuite de liquide parasitaire dans la circulation peut entraîner un choc anaphylactique. Finalement, c’est le kyste biliaire atypique qui pose le plus de problèmes (tableau 1), compte tenu de la fréquence élevée des kystes biliaires et la prise en charge radicalement différente pour le kyste biliaire et la TMK. Dans ce contexte, certains signes très évocateurs de TMK visibles au scanner et à l’IRM sont très utiles à connaître pour poser d’emblée une indication d’exérèse chirurgicale : une dilatation des voies biliaires en amont de la tumeur (mieux visible en IRM qu’au scanner), une prise de contraste des parois (figures 5E, 6E), et l’existence de nodules muraux (bourgeons charnus) (figure 5C). Ces signes sont détectables avec une bonne concordance inter-lecteurs et sont suffisamment prédictifs pour que la présence combinée de 2 ou 3 d’entre eux dans une seule lésion ait une spécificité de plus de 94 % pour le diagnostic de TMK (43). Inversement, la présence d’au moins 3 lésions kystiques supplémentaires ayant un aspect typique de kyste biliaire est un bon argument pour le diagnostic de kyste biliaire atypique plutôt que pour celui de TMK. Un autre signe particulièrement discriminant est la présence ou non d’une indentation au niveau de la base d’implantation des cloisons, associée à une convexité externe de la paroi du kyste de part et d’autre de chaque cloison (figure 6D). Dans l’étude princeps de Kovacs (48), ce signe était capable de discriminer à lui seul des kystes biliaires accolés d’une tumeur mucineuse kystique, avec une spécificité de 86 % pour le diagnostic de TMK (en l’absence d’indentation), et une spécificité de 91 % pour le diagnostic de kystes biliaires si les indentations sont visibles. Ces résultats ont été confirmés dans une seconde étude (49), où la spécificité de la présence d’indentations pour le diagnostic de kystes biliaires simples atteignait 100 %, alors que celle de leur absence n’avait une spécificité que de 56 % pour le diagnostic de TMK.
L’analyse du liquide intrakystique est en général déconseillée en cas de doute sur une TMK, a fortiori parvenue au stade de cystadénocarcinome, compte tenu d’un risque de dissémination de cellules tumorales le long du trajet de ponction. Néanmoins, deux intérêts peuvent être envisagés et ont été discutés dans la littérature : 1) le diagnostic différentiel entre une TMK et un kyste biliaire atypique et 2) le diagnostic différentiel entre une TMK bénigne et un cystadénocarcinome. Pour les tumeurs mucineuses kystiques pancréatiques, l’analyse du liquide a démontré son utilité pour identifier un cystadénocarcinome, principalement une concentration élevée de Ca 72-4. Cela n’a pas encore pu être démontré pour les TMK hépatiques compte tenu de la faible prévalence des formes dégénérées et la recommandation de ne pas ponctionner les TMK hépatiques. Dans la seule étude consacrée au sujet, qui avait inclus 114 malades opérés pour lesquels le diagnostic de la lésion kystique avait été confirmé avec certitude par l’anatomopathologique, seuls 4 cas de cystadénocarcinome avaient été inclus, ce qui n’avait pas permis de fournir de conclusion. En revanche, cette étude avait fourni des données utiles pour le diagnostic différentiel entre TMK et kystes biliaires. Dans les TMK, il existait une augmentation plus fréquente et plus marquée de l’ACE et du Ca 19-9, mais il n’était pas possible d’utiliser ces deux marqueurs pour discriminer TMK de kyste biliaire à l’échelon individuel, car des valeurs comparables d’ACE et de Ca 19-9 pouvaient s’observer dans ces deux lésions chez plus d’un tiers des malades. En revanche, le dosage du Ca 72-4 était très contributif : avec une valeur > 25 U/ml, il était possible de prédire un diagnostic de tumeur mucineuse kystique avec une sensibilité de 79 % et une spécificité de 97 % (AUROC : 0,98) (3).
Enfin, un outil sans doute intéressant mais non encore spécifiquement étudié pour l’analyse des tumeurs mucineuses kystiques intrahépatiques est le PET-scan au 18 FDG. On connaît en effet sa bonne rentabilité pour la détection des cystadénocarcinomes pancréatiques, histologiquement comparables aux cystadénocarcinomes hépatiques, et par conséquent son rôle clé pour déterminer la nature de la résection chirurgicale pancréatique. Pour la prise en charge des TMK hépatiques, toutes candidates à une même résection chirurgicale, le diagnostic préopératoire de cystadénocarcinome hépatique a, il est vrai, un enjeu beaucoup plus limité.
Traitement
Il n’existe actuellement aucune recommandation fondée sur les preuves concernant le traitement des TMK du foie, compte tenu de leur rareté. Le traitement recommandé par l’expérience est l’ablation chirurgicale de la tumeur, d’emblée, sans diagnostic invasif préalable ni traitement néo- adjuvant (50). La fenestration chirurgicale et les traitements percutanés exposent à un risque important (> 80 %) de récidive tumorale et doivent être proscrits. La résection, le plus souvent sous la forme d’une lobectomie, est la technique de référence et s’accompagne d’un excellent pronostic, avec une survie proche de 100 % à 5 ans et un taux de récidive inférieur à 10 %, qui serait favorisé par des manipulations tumorales antérieures à la résection (50). La résection n’est pas toujours possible lorsque la tumeur est trop volumineuse pour laisser un parenchyme hépatique sain suffisant.
On peut alors pratiquer une énucléation de la tumeur, avec des bons résultats post opératoires et à long terme, marqués par l’absence de récidive ou de transformation maligne, observées chez les seuls 40 patients traités de la sorte rapportés dans la littérature (51). On considère par conséquent qu’il n’y a pas de place pour la transplantation hépatique dans cette indication.
En cas de doute diagnostique entre une TMK bénigne et un kyste biliaire atypique persistant malgré l’imagerie, une attitude astucieuse pourrait être la réalisation d’un examen extemporané d’un fragment kystique, permettant de compléter le geste par une résection en cas de TMK, et par une fenestration en cas de kyste biliaire. Une série a rapporté le résultat de cette démarche chez 36 patients dont 33 avaient un diagnostic préopératoire de kyste biliaire et 3 de TMK bénigne (52). Les données de l’examen extemporané se sont toutes avérées fiables et ont permis de redresser le diagnostic chez 9 patients, 8 supposés atteints de kystes biliaires finalement traités par résection ou énucléation et 1 supposé atteint de TMK finalement traité par fenestration.
La question de la prise en charge postopératoire des cystadénocarcinomes, qui représentent moins de 8 % des TMK, n’est pas réglée. Dans la série de Vogt (46), 3 des 4 patients, chez lesquels un cystadénocarcinome avait été diagnostiqué sur la pièce d’exérèse, ont développé des métastases et 2 d’entre eux sont décédés dans les 12 mois. Il n’existe aucune thérapie ciblée pour les cystadénocarcinomes. Quelques rares expériences ont rapporté sous forme de cas clinique des patients atteints de formes non résécables ou métastatiques traités avec peu de succès par radiothérapie ou chimiothérapie, ou chimioembolisation artérielle. Dans une méta-analyse de 112 cas de cystadénocarcinome, Lauffer et al. ont rapporté une survie de 33 % à 5 ans pour les patients ayant une tumeur non résécable traitée par radiothérapie et chimiothérapie (53). Récemment, un cas clinique a rapporté l’observation d’une patiente traitée par l’apatinib, un agent anti VEGF, avec une absence de progression tumorale pendant 1 an (54). En raison du manque de données, aucune recommandation fondée sur les preuves ne peut être faite sur le rôle de la chimiothérapie et de la radiothérapie, et il n’existe pas de chapitre sur les cystadénocarcinomes dans le Thésaurus National de Cancérologie Digestive.
Tumeurs papillaires intracanalaires
Les tumeurs papillaires intracanalaires (TPI), anciennement nommées « papillomatose biliaire », sont caractérisées par une communication avec les voies biliaires et une dilatation des voies biliaires en aval due à la production de mucine luminale. Les TPI peuvent se développer n’importe où le long des voies biliaires. Elles ont un potentiel malin variable et sont classées en 5 stades : 1) TPI en dysplasie intraépithéliale de bas grade ou de grade intermédiaire, 2) en dysplasie de haut grade, 3) avec carcinome in situ, 4) avec envahissement microscopique, et 5) TPI associées à un carcinome invasif (21,43). Bien que l’étiopathogénie exacte de ces tumeurs ne soit pas encore complètement élucidée, on pense que l’inflammation chronique des voies biliaires secondaire à la maladie lithiasique, à la distomatose à Clonorchis sinensis, (fréquente en Asie), et au reflux de liquide pancréatique dans les voies biliaires (en cas de long canal commun bilio-pancréatique) pourrait être le facteur clé de la genèse et de l’oncogenèse des TPI (43).
Épidémiologie et présentation clinique
Les TPI sont très rares (moins de 300 cas rapportés), touchent les patients d’âge moyen, entre 40 et 70 ans, avec une légère prédilection pour le sexe masculin. Les pays d’Extrême-Orient (Japon, Corée, Taïwan) sont les plus touchés. Les TPI peuvent être asymptomatiques mais entraînent le plus souvent des douleurs de l’hypochondre droit, des épisodes récurrents d’angiocholite, ou un ictère obstructif intermittent. L’obstruction des voies biliaires peut être secondaire à la lithiase biliaire, fréquente, au détachement de débris tumoraux, ou à l’hypersécrétion de mucine.
Anatomopathologie
Macroscopiquement, les TPI se présentent sous la forme de masses papillaires friables, solitaires ou plus souvent multifocales touchant les voies biliaires intrahépatiques et/ ou extrahépatiques. Typiquement, ces lésions ont une sécrétion muqueuse et affichent une croissance lente. Microscopiquement, les cellules tumorales épithéliales biliaires présentent une prolifération papillaire très caractéristique avec de délicates tiges fibrovasculaires visibles dans les voies biliaires. En l’immunohistochimie, les TPI sont classés en quatre sous-types :pancréatobiliaire (le plus courant), intestinal, gastrique et oncocytaire (21,44).
Imagerie
En imagerie, les TPI sont classées en quatre types : type 1 : masse intracanalaire associée à une dilatation canalaire en amont ; type 2 : dilatation biliaire marquée sans masse visible ; type 3 : masse visible avec dilatation du biliaire en amont et en aval ; type 4 : dilatation kystique la voie biliaire avec masse papillaire (55). L’échographie peut détecter la dilatation biliaire et ses éventuelles conséquences, comme la présence de calculs, mais sa sensibilité est faible pour détecter les masses intracanalaires. Le scanner injecté décrit typiquement des masses intracanalaires prenant le contraste au temps artériel et se lavant au temps portal. Cette dernière caractéristique permet de discriminer les TPI des cholangiocarcinomes qui présentent en général un contingent fibreux qui entraînent une prise de contraste persistante et augmentant aux temps tardifs (43). La sensibilité du scanner pour la détection des TPI (50 %) reste néanmoins insuffisante. À l’IRM, les TPI formant une masse intracanalaire présentent une intensité beaucoup plus faible que celle de la bile en T2, ce qui facilite leur détection. L’IRM de diffusion pourrait favoriser la détection d’un carcinome invasif.
Les TPI de type 2 peuvent être difficiles à identifier au scanner et à l’IRM. Ceci est principalement dû au fait que la tumeur mucineuse est alors généralement isodense au scanner et isointense en IRM par rapport à la bile, ce qui la rend très difficile à détecter. Pour ces tumeurs, la cholangio- IRM est nécessaire pour détecter le « thread sign », un signe peu sensible mais hautement spécifique de TPI (Sp > 99 %), caractérisé par des stries hypointenses linéaires ou curvilignes intracanalaires dans les voies biliaires dilatées (56). Les TPI de type 4 peuvent être confondues avec des kystes biliaires simples, particulièrement lorsque la masse papillaire n’est pas visible ou passe inaperçue dans un kyste de grand volume. C’est la mise en évidence, en cholangio-IRM, d’une communication de la lésion kystique avec la voie biliaire, qui permet alors de rétablir le diagnostic. L’utilisation d’un produit de contraste hépatobiliaire (exemple : Multihanse®) peut aussi améliorer la détection de cette communication (43).
Il existe peu de données sur l’intérêt du PET-scan au FDG dans la prise en charge des TPI. Celui-ci pourrait être utile pour détecter une dysplasie de haut grade ou un carcinome invasif. La CPRE peut quant à elle détecter les tumeurs sous forme de défauts de remplissage irréguliers sur la paroi des voies biliaires et constater un écoulement de liquide mucineux par la papille. La cholangioscopie peut visualiser directement la partie solide d’une TPI, évaluer son extension dans les voies biliaires faire un brossage et des biopsies qui confirmeront le diagnostic (43).
Traitement
Compte tenu du potentiel malin des TPI, la résection chirurgicale est le traitement de choix. L’étendue de la résection chirurgicale dépend du degré d’atteinte des voies biliaires. Elle est surtout adaptée aux formes localisées. Un examen extemporané des marges de section doit être réalisé pour compléter si besoin la résection hépatique, l’étendre au cholédoque, ou réaliser une duodénopancréatectomie céphalique. Un curage ganglionnaire est indispensable. L’examen de la pièce d’exérèse pourra justifier d’un recours secondaire à la transplantation en cas de carcinome in situ ou invasif, à condition qu’il n’existe pas de ganglions envahis (21). Cette stratégie s’accompagne d’un risque significatif de récidive locale et d’apparition d’un cholangiocarcinome, qui justifient une surveillance à vie. Pour certains, la transplantation hépatique avec résection complète du cholédoque, voire duodénopancréatectomie céphalique, doit être envisagée d’emblée, même en l’absence d’épisodes angiocholitiques, car elle constitue le traitement « idéal » de ces tumeurs rares. Les résultats ne sont toutefois pas garantis, et l’évolution post transplantation restera défavorable en cas de tumeur invasive découverte sur l’explant. Cela peut justifier l’examen extemporané préalable d’une pièce de résection partielle de la lésion prédominante et des ganglions, qui contre-indiqueront la transplantation en cas d’envahissement ganglionnaire ou de carcinome invasif (57).
Lorsque la chirurgie est contre-indiquée, les alternatives thérapeutiques incluent le drainage biliaire percutané et les traitements cholangioscopiques locaux tels que la radiofréquence, l’ablation au laser et la coagulation au plasma d’argon. Dans l’ensemble, les TPI ont un meilleur pronostic que les cholangiocarcinomes des voies biliaires. Le taux de survie sans maladie à 5 ans serait de 81 % chez les patients ayant subi une résection chirurgicale curative. Cependant, des facteurs tels qu’une marge de résection chirurgicale positive, la présence d’adénopathies, une profondeur d’envahissement tumoral ≥ 5 mm et la présence de composants de carcinome invasif ≥ 10 % sont associés à un mauvais pronostic (43).
Kyste à revêtement cilié
Le kyste à revêtement cilié, lésion extrêmement rare, aussi appelée « kyste hépatique cilié congénital de l’intestin antérieur », est une lésion solitaire, généralement située dans la région sous-capsulaire du segment IV. Sa paroi est tapissée par un épithélium cylindrique pseudostratifié et cilié surmontant un tissu conjonctif, des fibres musculaires lisses et une capsule fibreuse périphérique. Il n’y a pas de canal biliaire. On suppose que ces kystes proviennent d’un bourgeonnement précoce et anormal de l’intestin primitif antérieur, à l’origine du foie. En imagerie, la lésion mesure moins de 4 cm et sa localisation typique doit faire d’emblée évoquer le diagnostic. Il peut exister à l’échographie un renforcement postérieur. Au scanner, la densité du kyste peut apparaître légèrement moins faible que celle d’un kyste biliaire simple. En IRM, le signal interne est de façon similaire légèrement plus hypointense en T2 et hyperintense en T1 par rapport au signal d’un kyste simple. Il n’existe jamais de réhaussement après injection de produit de contraste. Les kystes à revêtement cilié peuvent être symptomatiques en raison de leur localisation sous capsulaire. Ils sont décrits comme des lésions bénignes mais on a rapporté un potentiel de transformation maligne en carcinome épidermoïde, justifiant la réalisation une résection chirurgicale prophylactique lorsque le diagnostic est confirmé (1,43).
Kystes péribiliaires
Les kystes péribiliaires sont des dilatations kystiques macroscopiquement visibles des glandes peribiliaires (figure 7). Ils sont situés dans le hile hépatique et mesurent jusqu’à 1 cm de diamètre. Ils ont été décrits en association avec les maladies hépatiques polykystiques congénitales (figure 4E) et la cirrhose (58). Un nombre considérable de patients atteints de cirrhose et de pancréatite chronique alcoolique ont des kystes péribiliaires, et il a été suggéré que la maladie alcoolique du foie, tout comme son association avec une pancréatite chronique, sont des conditions favorisant
la formation des kystes péribiliaires, dont la présence serait étroitement corrélée à l’étendue de la fibrose pancréatique. La fibrose des glandes péribiliaires est en effet l’élément clé de la formation des kystes péribiliaires. Elle peut être secondaire à l’activation des cellules étoilées du foie mais serait surtout provoquée ou aggravée par une adénite alcoolo-induite qui induirait la fibrose cicatricielle des glandes péribiliaires. (59).
À l’imagerie, les kystes péribiliaires peuvent apparaître sous la forme d’amas de kystes discrets au niveau de la bifurcation portale, d’une chaîne de kystes simulant des voies biliaires anormales ou d’une structure tubulaire parallèle aux structures portes près du hile hépatique. Les kystes péribiliaires sont anéchogènes et ne présentent aucun flux détectable à l’examen Doppler. Au scanner, les kystes péribiliaires sont hypodenses sans aucune prise de contraste. En IRM (figure 6F), les kystes péribiliaires sont hyperintenses en pondération T2 et hypointenses en pondération T1, sans rehaussement après administration de gadolinium. Il existe le plus souvent des images caractéristiques de pancréatite chronique : atrophie pancréatique, calcifications parenchymateuses, et dilatation irrégulière des canaux pancréatiques. Il est important de différencier les kystes péribiliaires d’authentiques voies biliaires dilatées, en particulier dans le cadre d’une biliopathie portale, d’un œdème périportal, de tumeurs kystiques ou d’abcès afin d’éviter des interventions inutiles. En effet, les kystes péribiliaires ne se compliquent pas et ne dégénèrent pas.

Figure 7 : représentation schématique des glandes péribiliaires. D’après (58)
Tableau 3 : lésions hépatiques d’allure kystique| Lésions d’allure kystique | Principaux éléments d’orientation |
|---|
| Infectieuses : Abcès à pyogène, fongique, amibien, échinococcose alvéolaire | Contexte septique, sérologie parasitaire, examen microbiologique du liquide de ponction |
| Bilome | Contexte chirurgical (chirurgie hépato-biliaire) |
| Hématome | Contexte traumatique ou chirurgical |
| Pseudokyste | Pancréatite aiguë ; atteinte du lobe gauche |
| Endométriose | Jeune femme, douleurs périodiques de l’hypochondre droit |
| Grossesse extra-utérine intrahépatique | Jeune femme, douleurs de l’hypochondre droit, β-HCG ; atteinte du lobe droit. |
| Cancers primitifs : carcinome hépato-cellulaire, cholangiocarcinome, angiosarcome. | Hépatopathie chronique (cirrhose, cholangite sclérosante primitive), alphafœtoprotéine, exposition professionnelle |
| Métastases : tumeurs carcinoïdes, GIST, tumeur ovarienne, pancréatique, sarcome… | Contexte néoplasique ; cancer primitif connu, chimiothérapie |
| Hamartome mésenchymateux kystique | Enfant < 3 ans |
Lésions d’allure occasionnellement kystique
De nombreuses lésions, qui ne sont pas classées comme d’authentiques kystes, peuvent se présenter occasionnellement comme des kystes hépatiques (tableau 3). Pour la plupart d’entre elles, le contexte permet d’orienter facilement le diagnostic (1).
Tumeurs bénignes
Hémangiomes kystiques du foie
Certains hémangiomes peuvent rarement se présenter sous une forme kystique, et peuvent être difficiles à caractériser en imagerie lorsque le
rehaussement progressif après injection à l’imagerie en coupe, typique des hémangiomes, est mal visualisé.
Hamartomes mésenchymateux
Les hamartomes mésenchymateux sont les deuxièmes tumeurs hépatiques bénignes les plus fréquentes. Elles touchent l’enfant, typiquement avant l’âge de trois ans (43). L’architecture interne est variable : solide, kystique, ou mixte. Dans leur présentation kystique, les hamartomes mésenchymateux se présentent sous la forme d’une large lésion multiloculaire unique à l’imagerie. L’âge permet de différencier cette lésion du cystadénome, qui lui touche les femmes d’âge moyen.
Contexte traumatique ou chirurgical
Hématomes
Les hématomes sont généralement faciles à différencier des autres lésions kystiques par la connaissance d’antécédents, parfois anciens, de traumatisme ou d’intervention chirurgicale ou percutanée. Ils peuvent être en situation sous capsulaire ou en plein parenchyme hépatique. Selon leur stade d’évolution, ils peuvent apparaitre hyperdenses au scanner, hyperintenses en T1 à l’IRM, indiquant la présence de sang, mais ces caractéristiques peuvent disparaître avec le temps et la lésion se présenter comme hétérogène, voire comme un kyste hypodense au scanner (1).
Bilome
Le bilome est une collection intra ou péri-hépatique secondaire à un traumatisme spontané ou iatrogène des voies biliaires. Il s’observe surtout après chirurgie biliaire et apparaît comme une collection anéchogène bien limitée, située à proximité du pédicule hépatique ou de la vésicule biliaire. Il n’existe aucune prise de contraste au scanner (1). En cas de doute diagnostique, une ponction mettant en évidence un liquide riche en bilirubine aura l’avantage supplémentaire d’éliminer une surinfection et de prévenir un cholépéritoine.
Lésions spécifiques des jeunes femmes en âge de procréer
Endométriose hépatique
Le kyste d’endométriose est une lésion parenchymateuse ou sous capsulaire hépatique rare dont l’imagerie peut apparaître complexe avec un caillot nodulaire et septal et/ ou des produits de dégradation du sang. L’existence de douleurs épigastriques ou de l’hypochondre droit périodiques et la coexistence d’une autre localisation d’endométriose aident à faire le diagnostic (43).
Grossesse intrahépatique
Chez une femme jeune présentant une aménorrhée (mais celle-ci est inconstante) et des douleurs de l’hypochondre droit, une lésion kystique touchant typiquement le lobe hépatique droit peut correspondre au sac gestationnel d’une exceptionnelle grossesse extra-utérine intrahépatique.
Le fœtus, lorsqu’il est de taille suffisante, peut être parfaitement visible au scanner. Il est souvent encore vivant lors du diagnostic. L’intervention chirurgicale s’impose pour prévenir une rupture capsulaire hémorragique du foie lorsque la lésion est volumineuse, avec ou sans injection préalable de methotrexate pour interrompre la grossesse (60).
Contexte néoplasique
Plusieurs lésions néoplasiques primitives ou secondaires peuvent apparaître kystiques ou pseudokystiques. Cet aspect peut être la conséquence d’une différentiation kystique ou d’une nécrose intratumorale (1).
Tumeurs primitives
Parmi les tumeurs primitives, le carcinome hépatocellulaire et le cholangiocarcinome peuvent exceptionnellement se présenter comme des lésions kystiques, tout comme certains sarcomes primitifs du foie (1). L’aspect kystique peut s’observer après un traitement totalement efficace, (ex : séquestre post-thermoablation d’un carcinome hepatocellulaire) ou partiellement efficace, comme une chimioembolisation, une thérapie ciblée ou une chimiothérapie conventionnelle.
Métastases kystiques
Les métastatiques hépatiques d’allure kystique sont fréquentes et concernent plusieurs cancers primitifs : sarcomes, (liposarcome myxoïde et sarcome embryonnaire qui touche les enfants), tumeurs neuroendocrines (figures 6A, 6B), tumeurs stromales gastro-intestinales (GIST) (figure 6C), tumeurs mucineuses gastro-intestinales et ovariennes, et, bien sûr, les métastases de cystadénocarcinome pancréatique. 50 % des métastases hépatiques kystiques proviendraient d’un cancer colique (2). Les métastases kystiques sont le plus souvent multiples. Le signe radiologique le plus utile au diagnostic est la mise en évidence d’un rehaussement de paroi après injection de produit de contraste au scanner, en IRM ou à l’échographie de contraste, ce qui évoque d’emblée la nature tumorale des lésions (1,43). Lorsque le contexte néoplasique n’est pas connu, que la tumeur primitive n’est pas identifiée, l’examen histologique est indispensable et justifie une biopsie percutanée.
Références
- Vilgrain V. Lésions kystiques du foie. Gastroenterol Clin Biol 2001;25 :B167-B177.
- Rawla P, Sunkara T, Muralidharan M, Raj P. An updated review of cystic hepatic lesions. Clin Exp Hepatol 2019;5:22-29.
- Fuks D, Voitot H, Paradis V, Belghiti J, Vilgrain V, Farges O. Intracystic concentrations of tumour markers for the diagnosis of cystic liver lesions. Brit J Surg 2014;101:408-416.
- Bakoyiannis A, Delis S, Triantopoulou C, Dervenis C. Rare cystic liver lesions: a diagnostic and managing challenge. World J Gastroenterol 2013;19:7603-7618.
- Carrim ZI, Murchison JT. The prevalence of simple renal and hepatic cysts detected by spiral computed tomography. Clin Radiol. 2003;58:626-9.
- Precetti S, Gandon Y, Vilgrain V. Imagerie des lésions kystiques du foie. J Radiol 2007 ;88 :1061-1072
- Wijnands TFM, Gortjes APM, Gevers TJG, et al. Efficacy ans safety of aspiration sclerotherapy of simple hepatic cysts: a systematic review. Am J Radiol 2017;208:201-207.
- Benzimra J, Ronot M, Fuks D, et al. Hepatic cysts treated with percutaneous ethanol sclerotherapy : time to extend the indications to haemorrhagic cysts and polycystic liver disease. Eur Radiol 2014;24:1030-1038.
- Wijnands TFM, Ronot M, Gevers TJG, et al. Predictors of treatment response following aspiration sclerotherapy of hepatic cysts: an international pooled analysis of individual patient data. Eur Radiol 2017;27:741-748.
- Salame E. Quand opérer un kyste hépatique ? Hepato-Gastro et Oncologie Digestive 2019 ;26 :839-843.
- Gomez A, Wisneski AD, Luu HY, et al. Contemporary Management of Hepatic Cyst Disease: Techniques and Outcomes at a Tertiary Hepatobiliary Center. J Gastrointest Surg. 2021;25:77-84.
- Furumaya A, van Rosmalen BV, de Graeff JJ, et al. Systematic review on percutaneous aspiration and sclerotherapy versus surgery in symptomatic simple hepatic cysts. HPB 2021;23:11-24.
- Ettel M, Eze O, Xu R. Clinical and biological significance of precursor lesions of intrahepatic cholangiocarcinoma. World J Hepatol. 2015;7:2563-70.
- Guo Y, Jain D, Weinreb J. Von Meyenburg Complex: Current Concepts and Imaging Misconceptions. J Comput Assist Tomogr. 2019;43:846- 851.
- Panda N, Brackett D, Eymard C, et al. Liver Transplantation for Recurrent Cholangitis From Von Meyenburg Complexes. Transplant Direct. 2019;5:e428. doi: 10.1097/TXD.0000000000000867.
- Jain D, Sarode VR, Abdul-Karim FW, Homer R, Robert ME. Evidence for the neoplastic transformation of Von-Meyenburg complexes. Am J Surg Pathol. 2000;24:1131-9.
- Parekh V, Peker D. Malignant Transformation in Von-Meyenburg Complexes: Histologic and Immunohistochemical Clues With Illustrative Cases. Appl Immunohistochem Mol Morphol. 2015;23:607-14.
- Jain D, Ahrens W, Finkelstein S. Molecular evidence for the neoplastic potential of hepatic Von-Meyenburg complexes. Appl Immunohistochem Mol Morphol. 2010;18:166-71.
- Yang XY, Zhang HB, Wu B, Li AJ, Fu XH. Surgery is the preferred treatment for bile duct hamartomas. Mol Clin Oncol. 2017;7:649-653.
- Yonem O, Bayraktar Y. Clinical characteristics of Caroli’s disease. World J Gastroenterol. 2007;13:1930-1933.
- Antonini TM, Boillot O, Chiche L, et al. Les indications rares de transplantation hépatique. Hépato-Gastro et oncologie digestive 2021 ;28 :75-122.
- Santos-Laso A, Izquierdo-Sánchez L, Lee-Law PY, et al. New Advances in Polycystic Liver Diseases. Semin Liver Dis. 2017;37:45-55.
- Chauveau D. Polykystoses rénales et polykystoses hépatiques. Hépato-Gastro et oncologie digestive 2019 ;26 :830-838.
- Zhang ZY, Wang ZM, Huang Y. Polycystic liver disease: Classification, diagnosis, treatment process, and clinical management. World J Hepatol. 2020 27;12:72-83.
- Boerrigter Masyuk AI, Masyuk TV, Trussoni CE, Pirius NE, LaRusso NF. Autophagy Promotes Hepatic Cystogenesis in Polycystic Liver Disease via Depletion of Cholangiocyte Ciliogenic Proteins. Hepatology. 2022 (in press). doi: 10.1002/hep.32298.
- Ravine D, Gibson RN, Walker RG, et al. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994;343:824-827.
- MM, Bongers EMHF, Lugtenberg D, Nevens F, Drenth JPH. Polycystic liver disease genes: Practical considerations for genetic testing. Eur J Med Genet. 2021;64:104160.
- Barbier L, Ronot M, Aussilhou B, et al. Polycystic liver disease: Hepatic venous outflow obstruction lesions of the noncystic parenchyma have major consequences. Hepatology. 2018;68:652-662.
- Waanders E, van Keimpema L, Brouwer JT, et al. Carbohydrate antigen 19-9 is extremely elevated in polycystic liver disease. Liver Int. 2009;29:1389-1395.
- Sherstha R, McKinley C, Russ P, et al. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology. 1997;26:1282-1286.
- Aapkes SE, Bernts LHP, Barten TRM, et al. Estrogens in polycystic liver disease: A target for future therapies? Liver Int. 2021;41:2009-2019.
- Gu JY, Lu TF, Li QG, et al. Adult polycystic liver disease concomitant with severe anemia due to intracystic bleeding is an indication for liver transplantation: case report and review of literature. J Dig Dis. 2016;17:408-414.
- Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3’,5’-cyclic monophosphate. Gastroenterology. 2007;132:1104-1116.
- Temmerman F, Ho TA, Vanslembrouck R, et al. Lanreotide Reduces Liver Volume, But Might Not Improve Muscle Wasting or Weight Loss, in Patients With Symptomatic Polycystic Liver Disease. Clin Gastroenterol Hepatol. 2015;13:2353-2359.
- Munoz-Garrido P, Marin JJ, Perugorria MJ, et al. Ursodeoxycholic acid inhibits hepatic cystogenesis in experimental models of polycystic liver disease. J Hepatol. 2015;63:952-961.
- D’Agnolo HM, Kievit W, Takkenberg RB, et al. Ursodeoxycholic acid in advanced polycystic liver disease: A phase 2 multicenter randomized controlled trial. J Hepatol. 2016;65:601-607.
- Drenth JP, Chrispijn M, Nagorney DM, Kamath PS, Torres VE. Medical and surgical treatment options for polycystic liver disease. Hepatology. 2010;52:2223-2230.
- Chebib FT, Harmon A, Irazabal Mira MV, et al. Outcomes and Durability of Hepatic Reduction after Combined Partial Hepatectomy and Cyst Fenestration for Massive Polycystic Liver Disease. J Am Coll Surg. 2016;223:118-126.
- van Keimpema L, Nevens F, Adam R, et al. Excellent survival after liver transplantation for isolated polycystic liver disease: an European Liver Transplant Registry study. Transpl Int. 2011;24:1239-1245.
- Doshi SD, Bittermann T, Schiano TD, Goldberg DS. Waitlisted Candidates With Polycystic Liver Disease Are More Likely to be Transplanted Than Those With Chronic Liver Failure. Transplantation. 2017;101:1838-1844.
- Kirchner GI, Rifai K, Cantz T, et al. Outcome and quality of life in patients with polycystic liver disease after liver or combined liver-kidney transplantation. Liver Transpl. 2006;12:1268-1277. doi: 10.1002/lt.20780. PMID: 16741930.
- Coquillard C, Berger J, Daily M, et al. Combined liver-kidney transplantation for polycystic liver and kidney disease: analysis from the United Network for Organ Sharing dataset. Liver Int. 2016;36:1018-1025.
- Anderson MA, Bhati CS, Ganeshan D, Itani M. Hepatobiliary mucinous cystic neoplasms and mimics. Abdom Radiol 2021 (in press). doi: 10.1007/s00261-021-03303-5.
- Shyu S, Singhi AD. Cystic biliary tumors of the liver: diagnostic criteria and common pitfalls. Hum Pathol. 2021;112:70-83.
- Xu HX, Lu MD, Liu LN, et al. Imaging features of intrahepatic biliary cystadenoma and cystadenocarcinoma on B-mode and contrast-enhanced ultrasound. Ultraschall Med. 2012;33:E241-E249.
- Vogt DP, Henderson JM, Chmielewski E. Cystadenoma and cystadenocarcinoma of the liver: a single center experience. J Am Coll Surg. 2005;200:727-733.
- Quigley B, Reid MD, Pehlivanoglu B, et al. Hepatobiliary Mucinous Cystic Neoplasms With Ovarian Type Stroma (So-Called “Hepatobiliary Cystadenoma/Cystadenocarcinoma”): Clinicopathologic Analysis of 36 Cases Illustrates Rarity of Carcinomatous Change. Am J Surg Pathol. 2018;42:95-102.
- Kovacs MD, Sheafor DH, Burchett PF, Picard MM, Hardie AD. Differentiating biliary cystadenomas from benign hepatic cysts: Preliminary analysis of new predictive imaging features. Clin Imaging. 2018;49:44-47.
- Boyum JH, Sheedy SP, Graham RP, et al. Hepatic Mucinous Cystic Neoplasm Versus Simple Biliary Cyst: Assessment of Distinguishing Imaging Features Using CT and MRI. AJR Am J Roentgenol. 2021;216:403-411.
- Soares KC, Arnaoutakis DJ, Kamel I, et al. Cystic neoplasms of the liver: biliary cystadenoma and cystadenocarcinoma. J Am Coll Surg. 2014;218:119-128.
- Strasberg SM, Chapman WC. Enucleation of Biliary Cystadenomas: a Review. J Gastrointest Surg. 2021;25:2700-2706.
- Doussot A, Gluskin J, Groot-Koerkamp B, et al. The accuracy of pre-operative imaging in the management of hepatic cysts. HPB. 2015;17:889-95.
- Läuffer JM, Baer HU, Maurer CA, et al. Biliary cystadenocarcinoma of the liver: the need for complete resection. Eur J Cancer. 1998;34:1845- 1851.
- Yang Y, Mai W, Chen W, et al. Case Report: Low-Dose Apatinib in the Treatment of Intrahepatic Biliary Cystadenoma With Recurrence and Malignant Transformation. Front Oncol. 2021;11:676092.
- Park HJ, Kim SY, Kim HJ, et al. Intraductal Papillary Neoplasm of the Bile Duct: Clinical, Imaging, and Pathologic Features. AJR Am J Roentgenol. 2018;211:67-75.
- Hong GS, Byun JH, Kim JH, et al. Thread sign in biliary intraductal papillary mucinous neoplasm: a novel specific finding for MRI. Eur Radiol 2016;26:3112-3120.
- Vibert E, Dokmak S, Belghiti J. Surgical strategy of biliary papillomatosis in Western countries. J Hepatobiliary Pancreat Sci 2010;17:241-5.
- Katabathina VS, Flaherty EM, Dasyam AK, et al. “Biliary Diseases with Pancreatic Counterparts”: Cross-sectional Imaging Findings. Radiographics 2016;36:374-392.
- Matsubara T, Sato Y, Igarashi S, et al. Alcohol-related injury to peribiliary glands is a cause of peribiliary cysts: based on analysis of clinical and autopsy cases. J Clin Gastroenterol 2014;48:153-9.
- Wang J, Su Z, Lu S, et al. Diagnosis and management of primary hepatic pregnancy: literature review of 31 cases. Arch Gynecol Obstet 2018;298:235-242.