Mots-clés
Dyspnée, Hypertension artérielle pulmonaire, Syndrome hepato pulmonaire, Transplantation hépatique, Cardiomyopathie cirrhotique.
Liens d’intérêts
Aucun
Abréviations
- HTPoP : Hypertension PortoPulmonaire HTAP : Hypertension artérielle Pulmonaire
- PAPm : Pression Artérielle Pulmonaire moyenne PAPO : Pression Artérielle Pulmoniare d’Occlusion RVP : Resistances Vasculaires Pulmonaires
- ETT : Echocardiographie TransThoracique SHP : Syndrome Hepato-Pulmonaire
Introduction
La cirrhose peut être associée à des pathologies respiratoires variées et souvent méconnues en rapport avec une atteinte parenchymateuse, pleurale ou vasculaire. La dyspnée est un symptôme fréquent au cours de la cirrhose, notamment chez les patients en attente de transplantation hépatique. Après avoir exclu des pathologies pulmonaires ou cardiaques indépendantes de la maladie hépatique, il convient d’évoquer des causes en lien avec la maladie hépatique. Au cours de la cirrhose, la cause la plus fréquente de dyspnée est secondaire à la décompensation d’ascite, associée ou non à un épanchement pleural (hydrothorax). Deux pathologies distinctes, qui sont des maladies vasculaires pulmonaires, peuvent être associées à une hypertension portale (avec ou sans cirrhose) : le syndrome hépato-pulmonaire et l’hypertension porto-pulmonaire.
Par ailleurs il existe au cours de la cirrhose des modifications hémodynamiques pouvant être responsables d’une cardiopathie spécifique appelée cardiomyopathie de la cirrhose, dont la prise en charge n’est pas consensuelle.
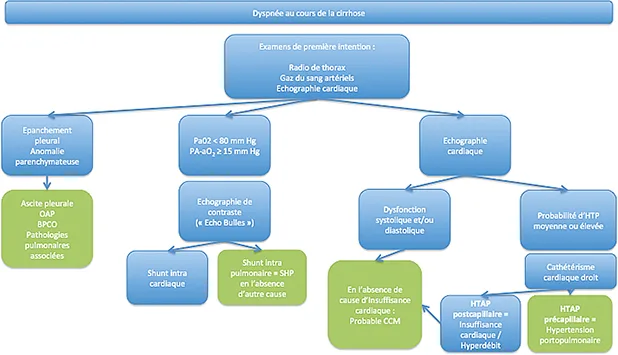
L’exploration d’une dyspnée en cas de cirrhose nécessite d’éliminer successivement tous ces diagnostics et peut être résumée dans un algorithme simple (figure 1).
Hypertension porto-pulmonaire
Définition
L’hypertension porto pulmonaire (HTPoP) est définie par l’association (a) d’une hypertension portale, (b) d’une élévation de la pression artérielle pulmonaire moyenne (PAPm) > 20 mm Hg, (c) d’une pression capillaire pulmonaire ou pression capillaire d’occlusion (PAPO) ≤ 15 mm Hg et (d) d’une élévation des résistances vasculaires pulmonaires (RVP) ≥ 3 UW (240 dynes.s.cm-5) définissant un hypertension pulmonaire pré- capillaire (1). L’HTPoP, comme toutes les formes d’hypertension artérielle pulmonaire, est caractérisée par un remodelage structurel et fonctionnel progressif des artères pulmonaires de petit calibre responsable de l’augmentation progressive des résistances vasculaires pulmonaires puis d’une défaillance ventriculaire droite. Les mécanismes impliqués dans la physiopathologie de l’HTPoP ne sont pas connus avec précision.
Signes cliniques, diagnostic et pronostic
Les signes cliniques de l’hypertension artérielle pulmonaire sont peu spécifiques et traduisent essentiellement les conséquences de l’augmentation des RVP sur la fonction ventriculaire droite. Ils se composent d’une dyspnée, de syncope ou une insuffisance cardiaque. On ne connaît pas de facteur prédictif de survenue de ce syndrome au cours de la cirrhose et la plupart des études suggèrent que la survenue et l’évolution de l’HTPoP sont indépendantes de la sévérité et de l’évolution de l’hypertension portale et de la maladie hépatique sous-jacente. Sa prévalence chez des patients candidats à la transplantation hépatique est comprise entre 2 et 5 % (2). Son impact sur le pronostic des patients justifie un dépistage systématique qui repose sur l’échographie cardiaque transthoracique (ETT) permettant de définir des seuils de probabilité d’hypertension pulmonaire (faible, intermédiaire ou élevé). L’ETT doit être répétée tous les 6 à 12 mois en attente de transplantation hépatique notamment pour ce dépistage.
Le cathétérisme cardiaque droit reste indispensable en cas de probabilité échographique intermédiaire ou élevée pour établir le diagnostic avec certitude et éliminer une augmentation des pressions pulmonaires liée au syndrome hyperkinétique ou à la rétention hydrosodée favorisés par la cirrhose (3). Le pronostic de l’HTPoP est mauvais avec une survie à 3 ans et 5 ans estimée à 69 % et 51 % respectivement. La survie dépend aussi bien de l’atteinte hémodynamique que de la sévérité de la maladie hépatique sous-jacente (4).

Figure 1 : Orientation diagnostique devant une dyspnée au cours de la cirrhose
Traitement
La prise en charge de l’HTPoP repose sur des mesures générales et l’utilisation des traitements ciblés de l’HTAP.
Le traitement non spécifique associera les diurétiques (spironolactone, furosémide) en cas de rétention hydrosodée. L’arrêt des bétabloquants est
souvent nécessaire du fait de leurs effets ionotrope et chronotrope négatifs, risquant d’aggraver la dysfonction ventriculaire droite.
L’utilisation des traitements spécifiques de l’HTAP dans la prise en charge de l’HTPoP se fonde sur les expériences cliniques et pharmacologiques acquises dans le traitement de l’HTAP idiopathique. Néanmoins, les données rétrospectives et observationnelles dont on dispose pour cette population de patients sont encourageantes en termes de tolérance et d’efficacité. Les trois classes thérapeutiques utilisées sont : les antagonistes
des récepteurs de l’endothéline, les inhibiteurs de la phosphodiestérase 5 et dans les cas les plus complexes les analogues de la prostaglandine. Dans tous les cas, l’initiation du traitement ainsi que son suivi doivent être réalisés dans les centres experts d’hypertension artérielle pulmonaire (centre de référence ou centres de compétences).
Antagonistes des récepteurs de l’endothéline
Un essai randomisé en double aveugle a inclus 85 patients à recevoir un antagoniste des récepteurs de l’endothéline (macitentan) ou un placebo dans le cadre d’une HTPoP. Dans le groupe traité par macitentan, il a été démontré une amélioration significative des paramètres hémodynamiques avec une diminution des RVP, une diminution de la PAPm et une amélioration de l’index cardiaque. Il n’existait pas d’amélioration des signes
fonctionnels. Le profil de tolérance était bon. Aucun patient n’était candidat à une transplantation hépatique (5). Il est à noter que ce traitement n’est pas encore disponible en France.
Le bosentan (antagoniste non sélectif des récepteurs A et B de l’endothéline 1) est un traitement approuvé dans les autres formes d’hypertension
pulmonaire. Son intérêt a été démontré dans une série rétrospective française, incluant 34 patients, dans l’HTPoP avec une diminution, voire une normalisation des RVP. Des cas d’augmentation des transaminases, voire d’aggravation de l’insuffisance hépatique, ont été décrits justifiant une surveillance de la fonction hépatique (6). Il est disponible en France.
L’ambisentan (antagoniste selectif des récepteurs A de l’endothéline 1) a montré des résultats comparables au bosentan en terme d’amélioration hémodynamique. Il semblerait qu’il existe moins de risques d’hépatotoxicité, en revanche son utilisation s’accompagne plus fréquemment d’une aggravation de la rétention hydrosodée (7).
Inhibiteurs de la phosphodiestérase 5 (IPDE5)
Le sildénafil et le tadalafil ont une activité antiproliférative et vascodilatatrice et sont utilisés dans différentes formes d’hypertension artérielle pulmonaire, mais aucune étude randomisée n’a évalué leur efficacité dans l’HTPoP. Il existe quelques études rétrospectives suggérant un intérêt du sildenafil chez les patients en attente de transplantation hépatique, et quelques cas cliniques concernant le tadalafil. L’utilisation des IPDE5 est peut- être plus aisée en l’absence d’hépatotoxicité ou de risque de rétention hydrosodée.
Analogues de la prostaglandine
L’effet bénéfique hémodynamique de l’époprosténol a été rapporté par plusieurs études rétrospectives, mais n’a jamais été étudié lors d’un essai contrôlé, prospectif, randomisé et son bénéfice en termes de survie n’est pas démontré. Son utilisation, nécessitant une perfusion continue, est réservée aux centres experts chez des patients très sélectionnés.
La transplantation hépatique
La transplantation hépatique n’est pas un traitement curatif de l’HTPoP, au contraire, celle-ci peut en être une contre-indication lorsque l’HTPoP est trop sévère et non contrôlée par le traitement médical. Les patients atteints d’HTPoP inscrits sur liste de transplantation ont un risque augmenté de mortalité. Ils peuvent bénéficier par conséquent d’une priorisation sur liste via une demande d’avis d’expert auprès de l’Agence de la Biomédecine. Au total, l’HTPoP est une complication rare mais particulièrement grave de la cirrhose. Son dépistage systématique (par l’échocardiographie cardiaque transthoracique) est recommandé chez les candidats à la transplantation car elle constitue un risque opératoire notable. (figures 2)
Syndrome hépato-pulmonaire
Le syndrome hépato-pulmonaire (SHP) est défini par l’association (a) d’une hypertension portale (avec ou sans cirrhose), (b) d’une augmentation de la différence alvéoloartérielle en oxygène (P(A-a)O2) calculée à partir d’une gazométrie prélevée en air ambiant en position assise et au repos (P(A-a)O2 ≥ 15 mm Hg ou ≥ 20 mm Hg chez des patients de plus de 64 ans), et (c) de dilatations vasculaires intrapulmonaires. La sévérité du SHP est appréciée en fonction du niveau d’hypoxémie, on parle de SHP léger en l’absence d’hypoxémie (PaO2 >80 mmHg), de SHP modéré lorsque la PaO2 est comprise entre 60 à 79 mm Hg, de SHP sévère lorsque la Pa02 est comprise entre 50 et 59 mm Hg et de SHP très sévère lorsque la PaO2 est inférieure à 50 mm Hg.
La prévalence du SHP est très variable selon les séries, estimée entre 5 et 50 % des patients. Cette disparité de prévalence est expliquée par la variabilité des critères diagnostiques utilisés et notamment les valeurs seuils pour l’hypoxémie. La prévalence des formes sévères (PaO2 < 60 mm Hg) semble être de l’ordre de 5 % (8).
Le SHP est consécutif à l’existence de dilatations vasculaires intra-pulmonaires. Il peut s’agir soit des dilatations capillaires excessives, soit de néovaisseaux. Les mécanismes physiopathologiques de ces dilatations vasculaires intra-pulmonaires sont incomplètement compris, ils font intervenir un certain nombre de médiateurs parmi lesquels le monoxyde d’azote, l’endothéline 1, le TNFα, et le monoxyde de carbone. Ces dilatations vasculaires intra-pulmonaires, entraînent en revanche une inadéquation dans le rapport ventilation/ perfusion.
Les zones perfusées sont ventilées mais la perfusion est trop importante par rapport à la ventilation, particulièrement chez les patients ayant un syndrome hyperkinétique, expliquant en partie l’hypoxémie. Cette hypoxémie est aggravée par la présence de shunts artério-veineux intra- pulmonaires, responsables d’un défaut d’oxygénation d’une partie du flux sanguin pulmonaire (8).
Les manifestations cliniques du SHP sont aspécifiques. Longtemps le SHP est asymptomatique, puis lorsque l’hypoxémie devient plus importante une dyspnée s’installe, et on peut observer un hippocratisme digital. La dyspnée est un signe non spécifique de SHP au cours de la cirrhose, il s’agit en effet d’un signe fréquent, pouvant traduire l’existence d’une ascite, d’un hydrothorax, d’une anémie, d’une dénutrition ou de maladies pulmonaires associées (BPCO par exemple). Au cours du syndrome hépato-pulmonaire, la dyspnée est caractérisée par une platypnée (dyspnée apparaissant lors du passage de la position couchée à la position debout), associée à une orthodéoxie (aggravation de l’hypoxémie en position debout). Cette dyspnée caractéristique est expliquée par la localisation préférentielle des dilatations vasculaires intrapulmonaires dans les bases pulmonaires. En effet, en position assise ou debout, ce sont les bases pulmonaires qui sont préférentiellement perfusées, diminuant ainsi le rapport ventilation/ perfusion et aggravant ainsi l’effet shunt.
Le dépistage du SHP est recommandé chez tous les patients candidats à la transplantation hépatique, cependant le dépistage par mesure de la saturation en oxygène n’est pas suffisamment sensible (9). Le diagnostic repose sur une augmentation de la différence alvéolo-artérielle en oxygène calculée sur des gaz du sang artériel réalisés en air ambiant. Le gaz du sang permet par ailleurs de grader la sévérité de l’hypoxémie. Il est nécessaire de mettre en évidence les conséquences des dilatations veineuses intra-pulmonaires. L’échographie cardiaque de contraste (ou échographie cardiaque aux micro-bulles) est l’examen de référence. Elle est réalisée en injectant par voie intraveineuse périphérique une solution saline qui aura été préalablement « secouée » pour générer des microbulles de 10 à 20 μm. Chez un sujet ne présentant pas de dilatations veineuses intra-pulmonaires, ces microbulles sont stoppées par le lit vasculaire pulmonaire. Ainsi, les bulles sont visualisées (sous l’aspect d’une opacification) après injection dans les cavités cardiaques droites mais ne le sont plus ensuite, ou très tardivement (après 9 à 10 cycles cardiaques) dans les cavités gauches.
L’apparition des microbulles après une période inférieure à 3 cycles cardiaques dans les cavités gauches évoque un shunt intra cardiaque (foramen ovale perméable par exemple). On parle de shunt intra-pulmonaire lorsque l’apparition des microbulles dans le cœur gauche se fait après 3 cycles cardiaques. L’échographie cardiaque transthoracique aux microbulles permet donc de mettre en évidence les conséquences des dilatations veineuses intra pulmonaires, mais ne permet pas de quantifier un éventuel shunt. La scintigraphie pulmonaire de perfusion aux macro- agrégats d’albumine marqués peut être utile pour quantifier le shunt. Elle ne permet pas cependant de différencier un shunt intra-cardiaque d’un shunt intra-pulmonaire. Une mesure quantitative de la distribution pulmonaire et cérébrale permet de calculer la fraction cérébrale correspondant aux agrégats passés à travers le système pulmonaire et donc de quantifier le shunt. Une fraction supérieure à 6 % permet de retenir le diagnostic de shunt droit gauche. La scintigraphie est moins sensible que l’échographie de contraste, en revanche, il existe une bonne corrélation entre la fraction du shunt et l’hypoxémie. La réalisation d’un angioscanner pulmonaire n’a pas d’intérêt pour le diagnostic de SHP, car les dilatations vasculaires sont très périphériques et souvent non visibles. En revanche, il permet de rechercher d’autres causes d’hypoxémie (ascite pleurale, atélectasie, pneumopathie infectieuse…) ou des comorbidités pulmonaires qui peuvent majorer l’hypoxémie.
Le SHP est un facteur de mauvais pronostic au cours de la cirrhose. Dans une étude rétrospective comparant des malades atteints de SHP à des malades atteints de cirrhose sans SHP, (appariée sur l’âge, l’étiologie, le score de Child et le score MELD), la survie était significativement plus faible en cas de SHP (23 % vs. 76 % p <0,001) (10). Dans cette même série, il a été constaté une aggravation de l’hypoxémie au cours du temps, avec en moyenne une perte de 5 mm Hg de PaO2 par an.
Aucun traitement médicamenteux ne permet d’améliorer les symptômes, le pronostic, ou l’histoire naturelle du syndrome hépato-pulmonaire.
L’oxygénothérapie est le seul traitement recommandé pour améliorer les symptômes associés à l’hypoxémie mais il n’améliore pas la survie des patients.
Le seul traitement curatif du SHP est la transplantation hépatique.
Figures 2 : Critères diagnostiques de l’hypertension artérielle pulmonaire
Figure 2.1 : Recommandations ESC/ ERS concernant la probabilité d’HTAP selon le flux d’insuffisance tricuspide [Vmax IT (m/s)] mesuré en ETT| Vmax IT (m/s) | Présence d’autres signes d’HTAP * | Probabilité échographique d’HTAP |
|---|
| ≤ 2,8 ou non mesurable | NON | FAIBLE |
| ≤ 2,8 ou non mesurable | OUI | MOYENNE |
| 2,9 – 3,4 | NON |
| 2,9 – 3,4 | OUI | ÉLEVÉE |
| > 3,4 | Non nécessaire |
* Les autres signes d’HTAP sont : ratio diamètre basale VD/VG > 1,0, aplatissement du septum interventriculaire, diamètre AP > 25 mm Hg, anomalie doppler pulmonaire, Diamètre VCI > 21 mm avec une diminution du collapsus inspiratoire, surface de l’OD > 18 cm2.
Figure 2.2 : Données hémodynamiques du cathétérisme cardiaque droit | PAPm | Index Cardiaque | RVP | Pression pulmonaire capillaire (PAPO) |
|---|
| HTAP post-capillaire (Syndrome hyperkinetique – Hypervolémie) | > 20 mm Hg | Augmenté (Sd hyperkinétique)
Normal (Hypervolémie)
Diminuée (Insuffisance cardiaque) | Diminuée
(< 3 UW) | Augmentée
(> 15 mm Hg) |
| HTAP pré-capillaire HTPoP | > 20 mm Hg | Variable | Augmentée
( ≥ 3 UW) | Normale
(< 15 mm Hg) |
PAPm = Pression Artérielle Pulmonaire moyenne, RVP : résistances vasculaires pulmonaires, PAPO : Pression artérielle pulmonaire d’occlusion
Cardiomyopathie du patient atteint de cirrhose
Au cours de la cirrhose, il existe des modifications hémodynamiques splanchniques et systémiques conduisant à une hypercinésie circulatoire caractérisée par une augmentation du débit cardiaque et une vasodilatation systémique. Malgré cette hypercinésie, l’adaptation cardiaque au stress (effort, épisode d’hémorragie digestive, sepsis) reste anormale et le concept de cardiomyopathie cirrhotique a été proposé en 2005. Elle est définie par une dysfonction cardiaque le plus souvent diastolique, associée à des anomalies électriques en l’absence de comorbidités cardiovasculaires.
De nouveaux critères diagnostiques ont été récemment proposés. Le diagnostic de cardiopathie cirrhotique peut être retenu lorsqu’il existe soit une dysfonction systolique soit une dysfonction diastolique en l’absence d’une cardiopathie d’autre cause. La dysfonction systolique est définie par une FeVG < 50 % ou des anomalies de la relaxation myocardique évaluées par strain échographie (doppler tissulaire). La dysfonction diastolique est définie par la présence d’au moins trois critères échographiques sur quatre habituellement utilisés pour le diagnostic de dysfonction diastolique, bien que la validité de ces marqueurs diastoliques reste débattue au cours de la cirrhose (11).
Les critères échographiques suggérant l’existence d’une dysfonction diastoliques sont les suivants : ratio E/e’ ≥ 15, volume oreillette gauche indexé > 34 ml/m2, onde e’ < 7 cm/sec, vitesse de régurgitation tricuspide > 2,8 m/sec.
En fonction des séries on estime que 40 à 60 % des patients atteints de cirrhose décompensée présentent une cardiopathie cirrhotique. Qu’il s’agisse d’un mécanisme adaptatif physiologique ou d’une cardiomyopathie spécifique de la cirrhose reste incertain. Il existe cependant un lien indéniable entre l’existence de modification cardiaque pathologique et la survenue d’une insuffisance rénale aiguë au cours de la cirrhose (12).
De manière beaucoup plus fréquente, il existe au cours de la cirrhose des comorbidités cardiovasculaires qui peuvent entraîner des dysfonctions cardiaques systoliques ou diastoliques. Quelle qu’en soit l’étiologie, la fonction cardiaque doit donc être évaluée chez des patients candidats à la transplantation hépatique ou à la mise en place d’un TIPS.
Lors d’un TIPS ou d’une transplantation hépatique il existe des modifications hémodynamiques (augmentation de la pré charge) qui peuvent entraîner une décompensation cardiaque si la fonction cardiaque n’a pas été correctement évaluée.
Récemment l’équipe de Toulouse a proposé un algorithme décisionnel, en utilisant des biomarqueurs courants (BNP ou NT-proBNP) ainsi que des données échographiques (données de fonction systolique ou diastolique) pour prédire le risque de survenue d’une décompensation cardiaque post TIPS (13).
Une évaluation cardiologique exhaustive est nécessaire avant la transplantation hépatique, celle-ci reste hétérogène en fonction des centres de transplantation.
La prise en charge de la cardiopathie du cirrhotique n’est pas consensuelle. En cas de décompensation cardiaque un traitement diurétique peut être mis en place. La place des bétabloquants n’est pas définie. Les inhibiteurs de conversion de l’angiotensine sont contre indiqués en cas de cirrhose décompensée car ils exposent à un risque d’insuffisance rénale.
Indication de transplantation hépatique Syndrome hépato-pulmonaire
Le seul traitement curatif du syndrome hépato-pulmonaire est la transplantation hépatique. Les échanges gazeux se normalisent entre 6 et 12 mois après la transplantation hépatique, mais ce délai peut parfois être prolongé, particulièrement chez les patients très hypoxémiques.
Du fait d’un mauvais pronostic, les patients en attente de transplantation pour un SHP justifient d’une dérogation au score d’attribution des greffons. Cette dérogation est justifiée chez les patients ayant une hypoxémie sévère (PaO2 < 60 mm Hg) (14). La survie des patients transplantés pour SHP est bonne, elle est de l’ordre de 70 à 80 % à 5 ans en fonction des séries. Certaines séries anciennes ont décrit une diminution de la survie post transplantation chez les malades les plus hypoxémiques, ceux ayant une PaO2 < 50 mm Hg. Plus récemment, une étude rétrospective sur près de 1 000 patients issus de la base de données de l’UNOS, a mis en évidence une surmortalité chez les malades qui présentaient une PaO2 < 44 mm Hg, comparés à ceux qui avaient une PaO2 comprise entre 44 et 54 mm Hg (15). Cette surmortalité pourrait faire discuter la contre-indication à la transplantation chez les malades les plus sévères. Cependant, lorsque la prise en charge post opératoire est optimisée, la mortalité reste acceptable (9 %) (16). Il n’existe pas, en France, de seuil inférieur de PaO2 devant contre indiquer la transplantation hépatique.
Hypertension porto-pulmonaire
L’HTPoP expose les patients candidats à une transplantation hépatique à un risque de défaillance ventriculaire droite en per et post-opératoire immédiat ainsi qu’à une aggravation de la pathologie vasculaire pulmonaire pendant plusieurs mois après la procédure. Une augmentation des résistances vasculaires pulmonaires semble être un facteur pronostic péjoratif (17). L’HTPoP non contrôlée représente donc une contre-indication à la TH. Il est clairement établi que la transplantation doit être contre-indiquée lorsque la PAPm excède à 50 mm Hg. Dans ce cas, le risque de décès péri-opératoire est majeur. Lorsque la PAPm est inférieure à 35 mm Hg et les RVP < 5 UW (400 dynes.s.cm-5), le risque de décès péri-opératoire semble comparable à celui des patients ayant une cirrhose non compliquée d’HTPoP et la transplantation peut être envisagée. Pour des valeurs de PAPm comprises entre 35 et 50 mm Hg, l’indication de transplantation hépatique est controversée et dépend du niveau de RVP et de la fonction ventriculaire droite. Elle ne semble pouvoir être envisagée que lorsque les résistances vasculaires pulmonaires sont inférieures à 3 UW (240 dynes.s.cm-5) (18).
Dans une étude rétrospective ancienne, il n’a pas été montré de sur-risque de décès lié à l’insuffisance ventriculaire droite chez les patients ayant une PAPm < 35 mm Hg ou comprise entre 35 et 50 mm Hg avec des RVP < 3 UW (18). Néanmoins, la pression pulmonaire n’est pas le seul facteur prédictif de décès. Les paramètres reflétant la fonction ventriculaire droite sont plus pertinents pour l’évaluation du risque chez des patients atteints d’HTAP. Ces données n’ont pas été clairement évaluées chez des candidats à la transplantation hépatique.
L’utilisation des traitements vasodilatateurs peut permettre d’obtenir des critères hémodynamiques compatibles avec la réalisation d’une transplantation hépatique.
L’amélioration hémodynamique par les vasodilatateurs avant transplantation permettait d’obtenir une survie après transplantation comparable à celle observée chez des malades atteints de cirrhose sans HTPoP (4,19). Chez des patients initialement en dehors des critères de transplantabilité, le traitement de l’HTAP permet de réduire la pression artérielle pulmonaire et d’améliorer la fonction ventriculaire droite afin d’amener les malades à la transplantation dans des conditions hémodynamiques acceptables chez environ 70 % d’entre eux (20).
Sur liste d’attente, il existe une augmentation de la mortalité en cas d’HTPoP, liée à la fois à la sévérité de la maladie hépatique et de l’état hémodynamique, justifiant une priorisation une fois que l’HTPoP est contrôlée (21).
En revanche, à distance de la transplantation hépatique, l’évolution de l’HTPoP est difficile à anticiper. Néanmoins, des données récentes ont montré que stabiliser, améliorer, voire guérir l’HTPoP, semble être un objectif réalisable en combinant traitements spécifiques de l’HTAP et transplantation chez des patients sélectionnés. Un sevrage des traitements vasodilatateurs semble en effet possible chez environ 20 à 40 % des patients (17,20). Les patients avec HTPoP et transplantés ont désormais une survie à long terme meilleure que les patients avec HTpoP non transplantés, quelle que soit la gravité de la maladie hépatique sous-jacente (4).
Le fait que l’HTPoP représente à elle seule une indication de transplantation hépatique est une stratégie évoquée mais qui reste actuellement controversée.
Conclusion
Les manifestations vasculaires pulmonaires au cours de la cirrhose sont représentées d’une part par le syndrome hépato-pulmonaire caractérisé par une hypoxémie, et dont le traitement repose sur la transplantation hépatique, et d’autre part par l’hypertension porto-pulmonaire caractérisée par une augmentation des pressions pulmonaires pouvant à terme aboutir à une insuffisance cardiaque droite. Cette hypertension porto-pulmonaire peut représenter une contre-indication à la transplantation hépatique. La cardiomyopathie du malade atteint de cirrhose est secondaire aux modifications hémodynamiques dues à l’hypertension portale dont les conséquences sont encore mal évaluées. Ces atteintes doivent être connues et systématiquement recherchées chez un cirrhotique, particulièrement en attente de transplantation hépatique.
Références
- Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019 Jan;53(1):1801913.
- Colle IO, Moreau R, Godinho E, Belghiti J, Ettori F, Cohen-Solal A, et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: a prospective study. Hepatology. 2003 Feb;37(2):401–9.
- Galiè N, Humbert M, Vachiery J-L, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Rev Esp Cardiol (Engl Ed). 2016 Feb;69(2):177.
- Savale L, Guimas M, Ebstein N, Fertin M, Jevnikar M, Renard S, et al. Portopulmonary hypertension in the current era of pulmonary hypertension management. J Hepatol. 2020 Mar 4;
- Sitbon O, Bosch J, Cottreel E, Csonka D, de Groote P, Hoeper MM, et al. Macitentan for the treatment of portopulmonary hypertension (PORTICO): a multicentre, randomised, double-blind, placebo-controlled, phase 4 trial. The Lancet Respiratory Medicine. 2019 Jul;7(7):594–604.
- Savale L, Magnier R, Le Pavec J, Jais X, Montani D, O’Callaghan DS, et al. Efficacy, safety and pharmacokinetics of bosentan in portopulmonary hypertension. European Respiratory Journal. 2013 Jan 1;41(1):96–103.
- Cartin-Ceba R, Swanson K, Iyer V, Wiesner RH, Krowka MJ. Safety and efficacy of ambrisentan for the treatment of portopulmonary hypertension. Chest. 2011 Jan;139(1):109–14.
- Rodríguez-Roisin R. Hepatopulmonary Syndrome — A Liver-Induced Lung Vascular Disorder. n engl j med. 2008;10.
- Forde KA, Fallon MB, Krowka MJ, Sprys M, Goldberg DS, Krok KL, et al. Pulse Oximetry Is Insensitive for Detection of Hepatopulmonary Syndrome in Patients Evaluated for Liver Transplantation. Hepatology. 2019 Jan;69(1):270–81.
- Swanson KL, Wiesner RH, Krowka MJ. Natural history of hepatopulmonary syndrome: Impact of liver transplantation. Hepatology. 2005 May;41(5):1122–9.
- Izzy M, VanWagner LB, Lin G, Altieri M, Findlay JY, Oh JK, et al. Redefining Cirrhotic Cardiomyopathy for the Modern Era. Hepatology. 2020 Jan;71(1):334–45.
- Ruiz-del-Arbol L, Monescillo A, Arocena C, Valer P, Ginès P, Moreira V, et al. Circulatory function and hepatorenal syndrome in cirrhosis. Hepatology. 2005 Aug;42(2):439–47.
- Billey C, Billet S, Robic MA, Cognet T, Guillaume M, Vinel JP, et al. A Prospective Study Identifying Predictive Factors of Cardiac Decompensation After Transjugular Intrahepatic Portosystemic Shunt: The Toulouse Algorithm. Hepatology. 2019 Dec;70(6):1928–41.
- Francoz C, Belghiti J, Castaing D, Chazouillères O, Duclos-Vallée J-C, Duvoux C, et al. Model for end-stage liver disease exceptions in the context of the French model for end-stage liver disease score-based liver allocation system. Liver Transpl. 2011 Oct;17(10):1137–51.
- Goldberg DS, Krok K, Batra S, Trotter JF, Kawut SM, Fallon MB. Impact of the Hepatopulmonary Syndrome MELD Exception Policy on Outcomes of Patients After Liver Transplantation: An Analysis of the UNOS Database. Gastroenterology. 2014 May;146(5):1256-1265.e1.
- Gupta S, Castel H, Rao RV, Picard M, Lilly L, Faughnan ME, et al. Improved survival after liver transplantation in patients with hepatopulmonary syndrome. Am J Transplant. 2010 Feb;10(2):354–63.
- Cartin-Ceba R, Burger C, Swanson K, Vargas H, Aqel B, Keaveny AP, et al. Clinical Outcomes after Liver Transplantation in Patients with Portopulmonary Hypertension. Transplantation [Internet]. 2020 Oct 12 [cited 2021 Jun 24];Publish Ahead of Print. Available from: https://journals. lww.com/10.1097/TP.0000000000003490
- Krowka M. Pulmonary Hemodynamics and Perioperative Cardiopulmonary-Related Mortality in Patients With Portopulmonary Hypertension Undergoing Liver Transplantation. Liver Transplantation. 2000 Jul;6(4):443–50.
- Raevens S, De Pauw M, Reyntjens K, Geerts A, Verhelst X, Berrevoet F, et al. Oral vasodilator therapy in patients with moderate to severe portopulmonary hypertension as a bridge to liver transplantation. Eur J Gastroenterol Hepatol. 2013 Apr;25(4):495–502.
- Savale L, Sattler C, Coilly A, Conti F, Renard S, Francoz C, et al. Long-term outcome in liver transplantation candidates with portopulmonary hypertension: Savale et al. Hepatology. 2017 May;65(5):1683–92.
- DuBrock HM, Goldberg DS, Sussman NL, Bartolome SD, Kadry Z, Salgia RJ, et al. Predictors of Waitlist Mortality in Portopulmonary Hypertension. Transplantation. 2017 Jul;101(7):1609–15.