Objectifs pédagogiques
- Savoir évoquer une cause rare de douleur abdominale chronique
- Connaître la démarche diagnostique
- Connaître les principes thérapeutiques
Toute reproduction ou réécriture, totale ou partielle, sans l’accord préalable écrit de la FMC HGE est interdite.
Conseil : Mayoly Spindler, Alnylam, SANOFI
Douleur abdominale ; Syndrome d’Ehlers-Danlos et hyperlaxité ; Narcotic bowel syndrome
Bien qu’il n’y ait pas de définition communément admise, on peut dire qu’une douleur abdominale est dite chronique ou persistante si elle est présente depuis au moins 6 mois, de manière continue ou intermittente et si elle résiste aux traitements habituels (1). Il s’agit d’une situation clinique fréquente rencontrée tant par les généralistes, que les gastro-entérologues ou les algologues mais aussi les chirurgiens, les gynécologues et sans doute les psychiatres. Dans la majorité des cas, la cause de ces douleurs abdominales chroniques est un syndrome fonctionnel comme le syndrome de l’intestin irritable (SII) ou la dyspepsie qui répondent à des critères diagnostiques précis, les critères de Rome IV (2,3). Dans certains cas, il existe une pathologie organique chronique sous-jacente connue comme une pancréatite chronique calcifiante, une maladie inflammatoire intestinale en rémission qui peut être retenue comme étant la cause de ces douleurs abdominales chroniques.
Chez certains patients, souvent avec des symptômes sévères, il faut savoir évoquer des causes rares ou inhabituelles de douleurs abdominales chroniques. Schématiquement il est possible de classer ces causes comme étant d’origine 1- digestive ou gynécologique, 2- systémique, 3- génétique, 4- dysautonomique ou centrale. Dans cette énumération, il ne faut surtout pas oublier les douleurs abdominales d’origine pariétale ou rhumatologique qui sont fréquentes et encore insuffisamment connues par le gastro-entérologue. Elles ne seront pas abordées dans cet article car détaillées lors de la FMC-HGE 2019 (Alcaraz P., Post’U 2019).
L’approche diagnostique chez un patient ayant des douleurs abdominales chroniques ou persistantes est basée, comme toujours, sur l’analyse des antécédents, l’historique des symptômes et leur description soigneuse (type de la douleur, localisation, irradiations, facteurs déclenchant/ calmant, relations avec l’alimentation, transit, symptômes associés digestifs et extra-digestifs, etc.), l’examen clinique, les examens biologiques et morphologiques déjà réalisés souvent dans un contexte d’errance diagnostique et de nomadisme médical. Dans cette situation un amaigrissement est presque constamment retrouvé, jusqu’à 85 % des cas (4), les patients faisant souvent des régimes très restrictifs dans l’espoir de diminuer leurs symptômes. Cet amaigrissement ne doit pas être considéré comme un signe d’alarme invitant à refaire un nouveau bilan morphologique. La recherche d’aliments déclenchant spécifiquement des symptômes doit être effectuée. L’efficacité, ou l’absence d’efficacité de régimes, comme un régime sans gluten ou un régime pauvre en FODMAPs, doit être recherchée. Compte tenu du caractère chronique des symptômes, toutes les explorations endoscopiques et morphologiques possibles ont le plus souvent déjà été réalisées et très souvent à plusieurs reprises. Même si cela est fastidieux, il est indispensable de revoir tous les examens et leurs résultats pour éviter de les répéter, à tort, ou à l’inverse pour en prescrire certains qui n’ont pas été réalisés ou dont les résultats sont douteux. Il est aussi indispensable de rechercher les antécédents chirurgicaux. Dans une cohorte de 103 patients avec douleurs abdominales chroniques, une intervention chirurgicale est rapportée comme un facteur déclenchant chez 27 % des patients et 68 % ont eu une ou plusieurs interventions chirurgicales sans aucun effet sur les symptômes (4). Il est par ailleurs bien établi que les patients ayant un SII ont un risque plus élevé d’avoir eu une cholécystectomie (RR 2,09 ; IC95 % 1,87-2,29), une appendicectomie (RR 1,45 ; IC95 % 1,33-1,56), une hystérectomie (RR 1,70 ; IC95 % 1,55-1,87) ou une chirurgie vertébrale essentiellement pour canal lombaire étroit (RR 1,22 ; IC95 % 1,05- 1,42) (5). Les informations sur les différents traitements reçus et leur efficacité doivent aussi être recueillies. De plus en plus souvent, les patients montrent les résultats de tests sérologiques avec dosages semi-quantitatifs d’Ig G à la recherche d’intolérances alimentaires. Chers (200 à 400 € environ) et non pris en charge par la CPAM, ces tests n’ont absolument aucune valeur scientifique, ne doivent pas être prescrits et leurs résultats ne doivent pas déboucher sur des régimes d’exclusion. De même, certains patients arrivent avec des résultats d’étude de microbiote. Peu sensibles et peu spécifiques, ces tests n’ont aucun intérêt et ne débouchent sur aucune prise en charge spécifique.
Cette analyse soigneuse est indispensable pour essayer d’orienter vers un diagnostic mais aussi pour identifier un éventuel facteur de décompensation (acute on chronic) ou dépister la survenue d’un cancer.
La GEE est une pathologie inflammatoire chronique caractérisée par une infiltration de la paroi digestive, parfois du péritoine, par des polynucléaires éosinophiles (PNE). Il s’agit d’une pathologie rare dont la prévalence est comprise entre 8,4 et 28 pour 10 (5) habitants (6). Les symptômes associent à des degrés divers : douleurs abdominales diffuses, nausées, vomissements, dyspepsie, diarrhée, altération de l’état général… L’association à une œsophagite à éosinophiles semble peu fréquente. La physiopathologie est mal comprise. Il existe un terrain atopique avec des allergies, en particulier alimentaire, un asthme, un eczéma dans seulement 50 % des cas environ. Une hyper-éosinophilie est inconstante. Le diagnostic repose sur l’augmentation du nombre de PNE dans la paroi digestive, justifiant la réalisation de biopsies systématiques en cas d’endoscopies normales, en se rappelant que la densité des PNE augmente tout au long du tube digestif. La normale chez l’adulte est < 20 PNE par champ dans le duodénum, < 40 dans le cæcum et < 50 dans le côlon gauche. Une consultation allergologique est indispensable si le diagnostic est retenu. Le traitement repose sur un régime d’éviction si une allergie alimentaire a été identifiée. Une corticothérapie est souvent nécessaire en cas de symptômes sévères. En l’absence d’étude randomisée, il est préconisé de faire un schéma type maladie inflammatoire intestinale (0,5 à 1 mg/kg/j durant 4 à 6 semaines, puis décroissance progressive) en sachant qu’il y a souvent une rechute en cas de décroissance trop rapide, plus ou moins associé à des antihistaminiques et au montélukast, un antagoniste des récepteurs aux leucotriènes. En cas de forme rebelle, l’utilisation de nouvelles biothérapies ciblant les interleukines spécifiques aux PNE comme le dupilumab, un anti-IL4, ou le mépolizumab, un anti-IL5, peut être discutée en centre expert hors AMM.
Diagnostiquée typiquement chez des patients après l’âge de 50 ans, la panniculite mésentérique est une masse inflammatoire de la racine du mésentère découverte de manière fortuite sur un scanner réalisée dans le cadre du bilan de douleurs digestives d’allure fonctionnelle. L’imputabilité de cette anomalie dans la genèse des symptômes est très discutée, seules les formes très inflammatoires associées à des adénopathies réactionnelles intenses sont peut-être responsable de douleurs (7). Il est classique de rechercher une néoplasie associée (mélanome, cancers urinaires, gynécologiques, colo-rectaux), un lymphome ou une tuberculose (8,9) au moment du diagnostic. Mais cette association est discutée (10) et dans la majorité des cas, le bilan est négatif. Il n’a pas été rapporté d’études de cohortes prospectives de bonne qualité, mais ces lésions ne semblent pas évolutives avec le temps. Il n’y a aucun traitement codifié.
L’IMC se définit comme un apport sanguin digestif insuffisant (11). L’IMC peut être occlusive ou non occlusive. La prévalence de l’IMC n’est pas connue, il est fréquent de retrouver une sténose athéromateuse des troncs mésentériques et/ ou cœliaque chez des patients âgés mais seulement une minorité des patients va rapporter des symptômes évocateurs. Les autres causes d’IMC occlusive sont des vascularites, les thromboses veineuses mésentériques et les causes d’IMC non occlusives sont toutes les causes de bas débit cardiaque. Le tableau clinique typique est une douleur abdominale postprandiale, survenant 10 à 20 min après la prise alimentaire et durant 1 à 2 heures. Les patients restreignent leur prise alimentaire avec une peur de manger. Un amaigrissement est fréquent (61-94 % des cas). Il peut s’y associer une diarrhée (19-61 %), des nausées (5-84 %), et une majoration de la douleur lors d’un exercice physique (43-76 %). L’examen clinique est le plus souvent normal, l’auscultation retrouve parfois un souffle abdominal. Il existe souvent un terrain athéromateux diffus. Le diagnostic repose sur le scanner et/ou l’IRM avec opacification aux temps artériels et veineux. Une sténose est considérée comme significative s’il y a une obstruction d’au moins 70 % d’un des troncs artériels. La prise en charge doit être effectuée en centre expert (au mieux une structure SURVI), associant gastro-entérologue, radiologue interventionnel et chirurgien vasculaire pour décider au cas par cas de la prise en charge adaptée (traitement médical, radiologie interventionnelle, chirurgie). Les risques étant de sur-traiter une sténose non significative ou à l’inverse de laisser en place une sténose significative avec un risque de survenue d’une ischémie aiguë mésentérique. Des guidelines très détaillés de l’UEG sur le diagnostic et la prise en charge de l’IMC sont disponibles depuis 2020 (11).
Il s’agit d’une compression du tronc cœliaque par le ligament arqué, un pont fibreux réunissant les 2 piliers du diaphragme, entraînant une ischémie occlusive. La réalité de ce syndrome est très discutée, une compression du tronc cœliaque par un ligament arqué étant retrouvée de manière fortuite chez 3 à 7 % des personnes ayant un scanner (11). Si une indication opératoire est retenue, le traitement est chirurgical avec souvent de bons résultats mais il n’y a pas d’études de cohortes prospectives.
Toutes les interventions chirurgicales abdominales et/ ou pelviennes sont susceptibles de générer des brides ou des adhérences post-opératoires quelle que soit la voie d’abord, laparotomie ou cœlioscopie. Chez un patient se plaignant de douleurs abdominales chroniques et ayant des antécédents chirurgicaux, en l’absence de phénomènes occlusifs, un diagnostic de douleurs adhérencielles est souvent évoqué. Il n’existe aucun examen complémentaire permettant de poser un diagnostic positif. De nombreuses études ouvertes non contrôlées avaient conclu qu’une adhésiolyse permettait de diminuer le syndrome douloureux. Cependant une étude randomisée hollandaise comparant adhésiolyse à une procédure fictive a montré qu’après un an (12) et 12 ans de suivis (13), les patients non opérés avaient moins de douleurs et moins de consommation de soins que ceux du groupe opéré.
La DSO est caractérisée par des douleurs abdominales aiguës récurrentes, avec des douleurs nocturnes insomniantes, de type biliaire ou pancréatique nécessitant de fréquents passages aux urgences (14). Un antécédent de cholécystectomie est fréquent. Il peut exister une élévation des transaminases au moment des crises. Après avoir éliminé toute suspicion de lithiase, le diagnostic peut être posé lors d’une CPRE, une manométrie du sphincter d’Oddi (geste morbide qui n’est plus jamais pratiqué) ou surtout une scintigraphie biliaire mais qui est difficile d’accès. Le diagnostic est souvent difficile, basé sur un faisceau d’argument. Le traitement est médical reposant essentiellement sur les antalgiques en cas de crises douloureuses. Depuis l’essai randomisé EPISOD (15), la sphinctérotomie endoscopique n’est plus recommandée compte tenu de son absence d’efficacité sur le syndrome douloureux et de sa morbidité.
L’endométriose est une maladie inflammatoire gynécologique touchant 7 à 10 % des femmes avant la ménopause (16,17). Par rapport à un groupe contrôle, les femmes ayant une endométriose signalent plus souvent une douleur abdominale diffuse, pas uniquement pelvienne, une constipation, des nausées et des ballonnements (17) et dans une méta-analyse récente le risque relatif d’avoir des symptômes de SII chez les femmes avec endométriose était de 3,26 (CI 95 % : 1,97-5,39) par rapport à des femmes sans endométriose (18). Chez les femmes en période d’activité génitale ayant des douleurs abdominales, une endométriose est maintenant très souvent évoquée mais, si cela n’a pas déjà été fait, elle doit être recherchée en réalisant les examens nécessaires (échographie, IRM) avant d’envisager une prise en charge spécialisée. Souvent, la douleur abdominale persiste malgré un traitement adapté ; il faut alors évoquer d’autres causes.
L’insuffisance surrénale peut être périphérique ou centrale et survient plus souvent entre 20 et 50 ans (19). La douleur abdominale est un des signes cliniques cardinaux associée à une fatigue, une faiblesse musculaire, un amaigrissement ou une hypotension. Ces signes cliniques, très peu spécifiques, peuvent, à tort, amener à poser un diagnostic de SII chez environ 30 à 40 % des patients (20). Chez des patients ayant une maladie inflammatoire intestinale ayant reçu une corticothérapie, une insuffisance surrénale est fréquente lors du sevrage (21) et ce diagnostic doit être évoqué en cas de douleurs abdominales persistantes. Chez des patients sous opiacés au long cours, la persistance d’une douleur abdominale peut être secondaire à une insuffisance surrénale dont le mécanisme est mal compris (22). Le diagnostic repose sur le dosage de la cortisolémie à 8 heures ou sur un test de stimulation par le tétracosactide (Synacthène®). En cas de diagnostic positif une supplémentation immédiate par hydrocortisone est recommandée pour éviter tout risque d’insuffisance surrénale aiguë, avant une prise en charge spécialisée.
La mastocytose systémique est une maladie hématologique caractérisée par l’accumulation de mastocytes dans différents organes comme le foie, la rate, la moelle osseuse et le tube digestif. Les symptômes sont peu spécifiques associant diarrhée, douleurs abdominales diffuses, symptômes de reflux, ballonnement, nausées, vomissements et sont très facilement confondus avec le SII entraînant un long retard au diagnostic, plus de 7 ans (23,24). Un antécédent d’ulcère gastrique ou duodénal peut éventuellement attirer l’attention mais aussi des manifestations extra-digestives comme un prurit, des antécédents de choc anaphylactique ou des réactions intenses lors de piqûres d’insectes, une pollakiurie, des épisodes de flush, une ostéoporose. Il existe souvent des signes neuro-psychiques avec des troubles de l’attention, de la concentration, une anxiété qui, eux aussi, ne sont pas spécifiques. Certains aliments libérant de l’histamine (alcools, crustacés, tomates, épices, blanc d’œuf, fraises, bananes, ananas, fruits exotiques, cacahuètes, noix, noisettes, chocolat) ou riches en histamine (vin, choucroute, fromages fermentés, charcuterie conserves, notamment de poisson et petits pois ou fruits de mer) peuvent favoriser l’apparition des symptômes. Le diagnostic de l’atteinte digestive est posé sur la présence en quantité anomale de mastocytes (> 15 par champ) sur les biopsies digestives après coloration spécifique, un infiltrat inflammatoire et parfois une atrophie villositaire partielle. Le dosage de la tryptasémie est en général élevé. La prise en charge repose sur un régime avec éviction du ou des aliments pouvant déclencher les symptômes, des anti-histaminiques et surtout les patients doivent être orientés vers des centres experts dans le cadre du réseau CEREMAST.
Contrairement à la mastocytose, le SAMA est caractérisé par l’absence d’anomalie quantitative des mastocytes, donc l’absence d’infiltration des tissus par des mastocytes. Le diagnostic est évoqué devant des symptômes similaires avec des douleurs abdominales diffuses d’allure fonctionnelle associées à des manifestations systémiques similaires comme flush, prurit, congestion nasale, enrouement, céphalées, palpitations qui ne sont absolument pas spécifiques. La tryptasémie est normale, une élévation de 2 µg/L ou de 20 % par rapport à la basale lors d’une crise sont des arguments évocateurs (25,26) ainsi que l’amélioration des symptômes par un traitement associant anti-H1, les moins sédatifs, anti-H2 (famotidine 40 mg x 2 par jour) et éventuellement du montélukast. Le traitement de seconde ligne repose sur le cromoglycate de sodium (100 à 400 mg par jour, préparation magistrale) (27). En cas d’échec, une prise en charge dans un centre de référence est justifiée. Très récemment, des données préliminaires suggèrent que le SAMA est fréquent chez des patients ayant un COVID long et pourrait expliquer certains symptômes digestifs tels que douleurs abdominales, diarrhée (28).
Une origine génétique de la douleur abdominale n’est pas fréquente mais peut être évoquée en cas d’antécédents familiaux évocateurs ou si le patient provient d’une zone géographique avec forte prévalence comme dans le cas de fièvre méditerranéenne.
Ce syndrome est la conséquence d’une mutation du gène de l’inhibiteur de la C1 estérase. Le type 1 est secondaire à une diminution quantitative de l’inhibiteur de la C1 estérase, le type 2 à une dysfonction de l’inhibiteur de la C1 estérase. Les symptômes les plus fréquents sont des œdèmes cutanés, des douleurs abdominales et au maximum un œdème laryngé qui est une urgence vitale (29,30). Les douleurs abdominales peuvent survenir par crises aiguës (en général durant 1 à 5 jours) ou bien se manifester par des douleurs abdominales chroniques qui sont présentes chez près de 80 % des patients. Les manifestations extra-digestives ne sont pas toujours présentes, ce qui rend le diagnostic compliqué. Le diagnostic est basé sur les antécédents familiaux, la recherche de facteurs déclenchant comme certains médicaments et sur l’imagerie abdominale (scanner, échographie) qui montre un épaississement des parois digestives et souvent une ascite avec la particularité que ces anomalies radiologiques fluctuent très rapidement, pouvant disparaître du jour au lendemain. Le diagnostic positif repose sur le dosage quantitatif et le dosage de l’activité de l’inhibiteur de la C1 estérase. Le traitement repose sur l’utilisation d’inhibiteurs de la C1 estérase en cas de poussée aiguë et sur des traitements d’entretien (androgènes, inhibiteur de la C1 estérase) en cas de symptômes sévères et récurrents.
Les porphyries hépatiques aiguës sont des maladies génétiques rares caractérisées par des crises dites neuro-viscérales et, dans certaines formes, des manifestations cutanées. La porphyrie aiguë intermittente (PAI) est la forme la plus fréquente avec une prévalence de la mutation relativement élevée (1/1 700) mais la pénétrance du gène est faible avec comme conséquence assez peu de sujets symptomatiques (31). Les crises surviennent essentiellement chez la femme entre 18 et 45 ans. La crise neuro-viscérale de la PAI est caractérisée par une douleur abdominale intense diffuse, pseudo-chirurgicale, une constipation, des signes neuropsychiques (asthénie, obnubilation, coma) parfois une atteinte sensitivomotrice des 4 membres. Le tableau biologique au moment de la crise montre simplement une hyponatrémie, avec comme diagnostic différentiel l’insuffisance surrénale aigue. Le diagnostic est posé par le dosage dans les urines des précurseurs de l’hème, l’acide delta aminolévulinique (ALA) et le porphobilinogène (PBG). Le diagnostic est souvent difficile avec une errance des patients durant de très nombreuses années (> 10 ans). Les facteurs déclenchants d’une crise aiguë sont le stress, l’alcool, les variations hormonales, un épisode infectieux, une prise médicamenteuse. Souvent, un patient ne fera qu’une seule crise dans sa vie. Quelques patients vont avoir des crises aiguës répétées plus ou moins espacées et une étude récente a montré qu’entre les crises, il persistait des douleurs abdominales chroniques non spécifiques associées à des nausées, une fatigue et une anxiété chez près de 65 % des patients (32). Il est donc possible que la fréquence de la PAI symptomatique soit sous-estimée, des études sont en cours pour essayer de préciser la prévalence de la PAI dans cette situation. Le diagnostic final repose sur la mise en évidence des anomalies génétiques chez le probant avec un dépistage chez les apparentés. Le traitement de la crise repose sur une hospitalisation, une réhydratation et des antalgiques. Le givosiran, un ARN interférentiel, a été récemment développé et a complétement changé la prise en charge des patients, réduisant la fréquence et l’intensité des crises de 75 % par rapport au placebo (33). Lorsqu’un diagnostic est suspecté, il ne faut pas hésiter à contacter le Centre Français des Porphyries (AP-HP Hôpital Louis Mourier) ou consulter le site www.porphyrie.net.
Le SED est un groupe hétérogène de maladies héréditaires du tissu conjonctif associant à des degrés divers une hypermobilité articulaire, une hyperélasticité cutanée et une fragilité tissulaire. La prévalence du SED est estimée à 1/5 000. De très nombreuses formes cliniques de SED ont été décrites. Les plus fréquents sont le SED classique, le SED hypermobile et le SED vasculaire.
SED classique (SEDc), SED hypermobile (SEDh), désordre du syndrome d’hypermobilité (DSH) :
Le SEDc et SEDh sont des maladies autosomiques dominantes, avec des mutations identifiées sur le gène codant le collagène de type V (COL5A1 ou COL5A2) pour le SEDc alors qu’aucune mutation n’a été identifiée pour le SEDh. Le diagnostic repose sur des critères majeurs, une hyper- extensibilité de la peau et une hyper-mobilité articulaire généralisée que l’on peut aisément rechercher à l’examen clinique à savoir : la dorsiflexion passive des auriculaires (5e doigt) au-delà de 90° ; l’apposition passive des pouces sur les fléchisseurs de l’avant-bras, l’hyper-extension du coude au-delà de 10°, l’hyper-extension du genou au-delà de 10°, la flexion du tronc vers l’avant avec les genoux complètement étendus de sorte que les paumes de main peuvent reposer à plat sur le sol. Ceci permet de calculer le score de Beighton (figure 1). Il est aussi possible d’utiliser un questionnaire simplifié qui permet d’orienter vers cette hypothèse diagnostique (tableau 1). Toute une série de critères mineurs permettent d’affiner le diagnostic (34). Il existe cependant un groupe de patients qui ne rentre pas dans ces critères stricts, ils sont maintenant classés dans le cadre des désordres du spectre de l’hyper-mobilité (DSH). Les symptômes digestifs fonctionnels sont extrêmement fréquents chez les patients SEDc/SEDh/ DSH. Dans une cohorte anglaise de 603 patients SED/DSH, 98 % des patients remplissaient les critères diagnostics de syndromes fonctionnels selon les critères de Rome IV (35). Dans cette cohorte, 75 % des patients avaient des douleurs abdominales chroniques au moins 1 jour par semaine durant les 3 mois précédents contre 14 % dans le groupe contrôle. Au sein d’une cohorte française, essentiellement des patients avec SEDh, nous avons montré que 48 % remplissaient les critères diagnostics du SII, 36 % celui de constipation fonctionnelle et 76 % celui de reflux avec un très fort retentissement sur la qualité de vie (36). Enfin, en 2022 nous avons confirmé ces résultats dans une cohorte de patients SEDc, SEDh et DSH avec des symptômes plus sévères dans le groupe DSH (Placide PA et al., JFHOD 2022). Il n’y aucune explication rationnelle permettant d’expliquer la très grande fréquence des douleurs abdominales chroniques dans cette population. Elles sont souvent associées à des douleurs musculaires et articulaires diffuses avec des troubles de la proprioception, parfois une dysautonomie. Une étude récente a montré qu’il existait une hypo-sensibilité rectale à la distension chez des patients SEDh/HSD constipés par rapport à un groupe contrôle, sans augmentation de troubles de la statique pelvienne ni allongement du temps de transit colique (37). Cette anomalie permet d’expliquer la constipation, ne permet pas de comprendre l’origine des douleurs abdominales. La prise en charge diagnostique de ces patients est souvent complexe. Une endoscopie, si elle est nécessaire, peut être réalisée sans risque majeur de complications chez des patients SEDc/SEDh/DSH (38). De manière ponctuelle, ces douleurs d’allure fonctionnelle peuvent être associées à des troubles de la motricité œsophagienne ou une gastroparésie. La prise en charge thérapeutique est compliquée chez ces patients très symptomatiques, aucune étude randomisée n’a été rapportée. La prise en charge de la douleur abdominale doit s’intégrer dans une prise en charge multidisciplinaire au mieux coordonnée par un centre expert dans le cadre du réseau des SED non vasculaires assurant le diagnostic et la coordination des soins pour les patients atteints de SED/DSH (filière OSCAR, maladies rares de l’os, du calcium et du cartilage : https://filiere- oscar.fr). Le traitement doit être pragmatique et repose sur les antispasmodiques, les antalgiques de niveau 1 ou 2, les régulateurs du transit, les IPP. Quelques données suggèrent un effet bénéfique du régime sans gluten (39).
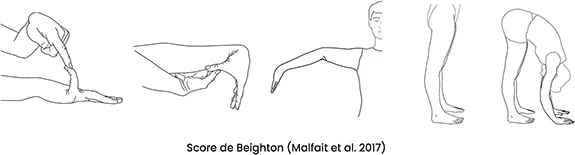
Figure 1 : Manœuvre pour calculer le score de Begihton
| 1. | Pouvez-vous aujourd’hui (ou avez-vous déjà pu) poser vos mains à plat sur le sol sans plier les genoux ? |
| 2. | Pouvez-vous maintenant (ou avez-vous déjà pu) plier votre pouce pour toucher votre avant-bras ? |
| 3. | Enfant, amusiez-vous vos amis en contorsionnant votre corps dans des positions étranges, ou pouviez-vous faire le grand écart ? |
| 4. | Enfant ou adolescent, votre épaule ou votre genou se sont-ils luxés plus d’une fois ? |
| 5. | Vous considérez-vous avec des articulations hyperlaxes ? |
Les manifestations digestives du SED vasculaire sont davantage centrées sur des problèmes de perforations spontanées ou d’hémorragie digestive que des douleurs abdominales chroniques (40). Compte tenu du risque très élevé de perforation, l’indication d’une endoscopie doit être très réfléchie et sa réalisation effectuée avec les plus grandes précautions.
La FMF est une maladie monogénique, c’est la maladie auto-inflammatoire la plus fréquente. Elle a été décrite dans la population du pourtour méditerranéen où la prévalence est très élevée, environ 20 pour 100 (41). Elle est caractérisée par des poussées inflammatoires touchant les séreuses associées à de la fièvre et un syndrome inflammatoire biologique marqué avec une élévation importante de la CRP. La douleur abdominale est intense avec à l’examen un abdomen pseudo-chirurgical au moment des poussées. Souvent, le patient a eu une ou plusieurs cœlioscopies exploratrices non contributives avant que le diagnostic ne soit posé. Typiquement les symptômes durent quelques jours, les signes cliniques disparaissent et le syndrome inflammatoire se corrige. En dehors des crises, le bilan est strictement normal. Le diagnostic repose sur la mise en évidence d’une mutation sur le gène MEFV. Cependant plus de 300 variants ont été identifiés dans la littérature et les panels de tests génétiques utilisés ne recherchent que les 6 à 8 mutations les plus fréquentes. Le traitement repose sur la colchicine à la dose de 1 à 2 mg qui permet de réduire la fréquence et l’intensité des crises et met à l’abri de la survenue d’une amylose, principale complication à long terme. En cas de doute diagnostique fort, si les tests génétiques sont négatifs, il est préconisé de faire un traitement d’épreuve par colchicine pendant quelques mois.
D’autres maladies auto-inflammatoires, beaucoup plus rares, comme le TRAPS syndrome ou le déficit en mévalonate kinase associent douleurs abdominales et fièvre mais les épisodes d’hyperthermies sont en général plus longs (2-4 semaines).
Le LPAC syndrome est associé à une mutation du gène ABCB4 qui code la protéine multidrug resistance 3 (MDR3). Une anomalie sur le fonctionnement de la MDR3 réduit la concentration biliaire de phosphatidylcholine, diminue la solubilisation du cholestérol et aboutit à la formation de lithiase. Le LPAC syndrome est retrouvé chez environ 1 % des patients ayant une lithiase (42). Typiquement, il s’agit de patients ayant des douleurs biliaires récurrentes après une cholécystectomie. Le diagnostic est fait à l’échographie avec l’aspect typique en queue de comète. Le diagnostic repose sur la présence d’au moins 2 des 3 critères suivant : 1- apparition avant l’âge de 40 ans ; 2- récidive des symptômes après une cholécystectomie ; 3- aspect en queue de comète intrahépatique à l’échographie. Le traitement repose sur l’acide ursodésoxycholique qui, en règle générale, entraîne une amélioration rapide des symptômes.
Le STOP est défini par la présence d’une tachycardie à l’orthostatisme (augmentation du rythme cardiaque > 30 min après 10 min d’orthostatisme, sans hypotension) depuis au moins 6 mois (43). Le STOP est souvent responsable de symptômes généraux tel éblouissement, vision floue, céphalées. Les symptômes digestifs sont très fréquents associant douleurs abdominales chroniques non spécifiques (70-80 %), nausées, vomissements, ballonnements, constipation (44). La physiopathologie des symptômes digestifs au cours du STOP est inconnue, le rôle d’une dysautonomie est fortement suspecté. Ce syndrome ayant été identifié assez récemment, il n’y a aucune étude randomisée en règle générale et pour la prise en charge des symptômes digestifs en particulier (45).
Compte tenu du chevauchement de symptômes, de leur manque de spécificité et des difficultés à aboutir à un diagnostic positif (à l’exception du STOP), une association entre SAMA, SEDh et STOP a parfois été évoquée mais les résultats des études sont discutables (46). Récemment, une étude de cohorte anglaise menée chez 616 patients SED/HSD a montré que 37 % des patients avaient un diagnostic de STOP. Ce sous-groupe de patients avait plus souvent des symptômes digestifs d’allure fonctionnelle souvent sévères avec un fort impact sur la qualité de vie (47).
Le narcotic bowel syndrome est caractérisé par la survenue et l’aggravation de douleurs abdominales chez des patients prenant des opioïdes de manière chronique avec comme conséquence une augmentation des doses de morphiniques qui provoque une augmentation des symptômes digestifs (48). La douleur abdominale est souvent sévère à très sévère et doit survenir de manière quotidienne depuis au moins 3 mois. Il y a souvent d’autres symptômes digestifs associés conséquences de la prise chronique de morphinique tels que nausées, vomissements, constipation, ballonnements. On estime qu’environ 5 % des patients sous morphiniques au long cours développent un narcotic bowel syndrome. Le mécanisme physiopathologique principal est l’induction d’un phénomène d’hyperalgésie centrale par la morphine. La prise en charge thérapeutique comprend une part importante d’éducation thérapeutique en expliquant aux patients le rôle néfaste des morphiniques, une rotation des molécules avec une décroissance progressive pour essayer d’arriver à un sevrage associé à des antidépresseurs et/ ou des anxiolytiques. Les molécules antagonistes des récepteurs périphériques de la morphine (PAMORA), tel que le naloxégol, ne semblent pas utiles pour gérer la composante douloureuse.
Ces dernières années, le SDAFC a été identifié comme un syndrome fonctionnel spécifique. Il répond à des critères diagnostics précis selon la classification de Rome IV (tableau 2) (49). La douleur doit être chronique, non feinte, non reliée aux événements physiologiques habituels comme le repas ou la défécation et les critères diagnostiques du SII ne doivent pas être présents. Finalement, un grand nombre de patients avec des douleurs abdominales chroniques rentrent dans ce cadre diagnostique. Les symptômes sont souvent sévères, toujours sources d’un nomadisme médical et de répétition d’examens. Le traitement repose essentiellement sur les médicaments avec action centrale comme les antidépresseurs tricycliques ou la duloxétine en première intention à dose progressive (4). Les associations de 2 ou plusieurs molécules à action centrale semblent plus efficaces que les monothérapies, comme toujours il s’agit d’avis d’experts en l’absence d’études randomisées. Il est important d’expliquer aux patients que ces molécules sont utilisées pour leur action antalgique et non pour leur action sur les troubles de l’humeur, même s’il existe souvent un terrain anxieux ou dépressif associé. De même, il est aussi important d’expliquer aux patients les différents effets secondaires ce qui sera source d’une meilleure observance et, on peut l’espérer, d’efficacité.
| Douleur abdominale continue ou presque |
| Sans relations avec événements physiologiques : – alimentation, défécation, cycle menstruel |
| Retentissement sur les fonctions quotidiennes : – scolarité, travail, vie personnelle |
| Douleur non feinte |
| Absence de critères des autres syndromes fonctionnels, absence d’anomalie structurelle |
| Présence durant les 3 derniers mois, apparition 6 mois avant le diagnostic |
Il n’est pas possible de définir un algorithme de prise en charge spécifique chez les patients consultant pour des douleurs abdominales chroniques (figure 2). L’analyse sémiologique des symptômes est un point essentiel car il n’est pas raisonnable d’envisager systématiquement toutes les causes de douleurs abdominales inhabituelles. Schématiquement, chez des patients ayant des douleurs abdominales chroniques ne rentrant pas dans le cadre diagnostic du SII ou de la dyspepsie, on peut dire :
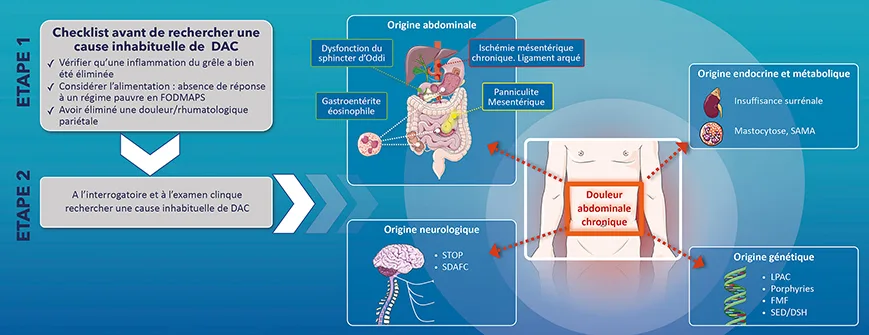
Figure 2 : Douleurs abdominales chroniques (DAC) : causes inabituelles
Il ne faut pas hésiter à émettre de nouvelles hypothèses diagnostiques à mesure que le temps passe et que le tableau sémiologique évolue. Et, même si ce n’est pas l’objet de cette mise au point, il ne faut jamais oublier les douleurs abdominales d’origine pariétale ou rhumatologiques.
ALA : acide delta amino lévulinique
DSH : désordres du spectre de l’hypermobilité DSO : dysfonction du sphincter d’Oddi
FMF : fièvre méditerranéenne familiale
FODMAPS : fermentable oligo-, di-, and monosaccharides and polyols GEE : gastroentérite à éosinopiles IMC : ischémie mésentérique chronique
LPAC : low phospholipid associated cholelithiasis
PAI : porphyrie aiguë intermittente
PBG : porphobolinogène
PNE : polynucléaires éosinophile
SAMA : syndrome d’activation mastocytaire
SDAF : syndrome de douleur abdominale fonctionnelle
SDAFC : Syndrome de Douleurs Abdominales Fonctionnelles Centrales SED : syndrome d’Ehlers Danlos
SEDc : Syndrome d’Ehlers-Danlos classique SEDh : Syndrome d’Ehlers-Danlos hypermobile SII : syndrome de l’intestin irritable
STOP : syndrome de tachycardie orthostatique posturale
FMC HGE : Organisme certifié Qualiopi pour la catégorie ACTIONS DE FORMATION.
