Liens d’intérêt
Aucun conflit d’intérêt en lieu avec la thématique
Mots-clés
Coéfficient de saturation de la transferrine, hémochromatose, hyperferritinémie dysmétabolique, IRM
Introduction
L’hyperferritinémie est un motif très fréquent de consultation en hépato-gastroentérologie. Il existe de nombreuses causes d’hyperferritinémie. Une bonne connaissance du métabolisme du fer, des différentes causes et des outils diagnostics non invasifs est nécessaire pour aboutir au diagnostic étiologique et ainsi permettre une prise en charge optimale. Des progrès considérables ont été réalisés ces 20 dernières années dans la compréhension du métabolisme du fer (1). Cette meilleure connaissance permettra très probablement, dans les années à venir, le développement de nouveaux outils diagnostiques et thérapeutiques.
Rappel physiologique
Le fer participe au transport de l’oxygène, à la synthèse de l’ADN et de l’ATP. Le stock en fer de l’organisme est d’environ 3 à 4 g chez l’homme et varie en fonction des périodes de la vie génitale chez la femme. Les apports quotidiens de fer compensent les pertes digestives et cutanées qui sont d’environ 1 à 2 mg/j. L’érythropoïèse consomme 25 à 30 mg de fer par jour qui lui est fourni par le recyclage du fer provenant des globules rouges sénescents qui sont phagocytés par les macrophages tissulaires en particulier au niveau du foie et de la rate (2). Une faible partie du fer circule dans le plasma lié à la transferrine (2 à 3 mg). En raison de son rôle physiologique majeur et de ses capacités toxiques en cas de surcharge en fer, il est indispensable que le fer soit parfaitement régulé. En cas de surcharge en fer, quand le coefficient de saturation dépasse 70 à 80 %, on observe l’apparition du fer non lié à la transferrine (NTBI = non transferrin bound iron) toxique en raison de sa capacité à générer des radicaux libres après pénétration dans les cellules en particulier hépatiques, sans passer par les récepteurs à la transferrine (3). La proportion de fer qui pénètre dans les cellules hépatiques est appelé LPI (labile plasma iron) ou pool labile plasmatique (4).
Le fer est absorbé au niveau digestif grâce au transporteur DMT-1 (DiMetal Trasporter 1). Le fer est ensuite exporté par la ferroportine au niveau basal puis transformé en fer ferrique (Fe3+) par l’hephaestine avant de se lier à la transferrine. Il existe une régulation du métabolisme du fer au niveau cellulaire et au niveau systémique. Au niveau cellulaire, deux systèmes sont responsables de la régulation du fer : le facteur transcriptionnel sensible à l’hypoxie (HIF2alpha) et les protéines régulatrices du fer (IRP/IRE) (1). Au niveau systémique, l’hormone qui contrôle l’absorption du fer est l’hepcidine. Elle est principalement synthétisée par les hépatocytes. Elle est responsable d’une inhibition de la synthèse de DMT-1 et d’une internalisation de la ferroportine qui est ensuite dégradée dans les lysosomes (1) au niveau des hépatocytes, des macrophages et de l’intestin. Le métabolisme du fer est également régulé par l’érythropoïèse. Quand l’érythropoïèse est stimulée par une hémorragie et/ ou une hypoxie, on observe une diminution importante de l’hepcidine. Plusieurs molécules sont candidates au rôle de régulateur érythropoïétique du métabolisme du fer. Actuellement le candidat le plus sérieux semble être l’ERFE (érythroferrone) (5). La boucle régulatrice est stimulée par l’EPO qui entraîne une prolifération des progéniteurs médullaires des globules rouges qui synthétisent l’ERFE (6) inhibant ainsi la synthèse d’hepcidine. Ce mécanisme explique la surcharge en fer par hyperabsorption digestive observée par exemple dans les thalassémies non dépendantes des transfusions ou les syndromes myélodysplasiques (7).
Principales causes d’hyperferritinémie
Cytolyse sans surcharge en fer
Il existe très peu de données de qualité dans la littérature sur la fréquence des anomalies du bilan du fer (CST et Ferritine) aussi bien pour les cytolyses hépatiques que pour les cytolyses musculaires (8). Le coefficient de saturation est soit normal, soit augmenté. La ferritine peut dépasser 20 000 ng/mL dans les cytolyses aiguës. La recherche d’une atteinte musculaire fait appel à l’interrogatoire et au dosage des CPK.
Inflammation sans surcharge en fer
L’inflammation est une des principales causes d’hyperferritinémie (1). La ferritine peut dépasser 50 000 ng/mL. Les causes d’inflammation sont multiples. L’inflammation peut être présente sans augmentation de la CRP et une anémie inflammatoire peut être associée. Dans un quart à la moitié des cas, l’anémie inflammatoire s’accompagne d’une anémie par carence en fer. En réponse à la présence de molécules microbiennes, d’antigènes tumoraux ou d’auto-antigènes, l’organisme fabrique de nombreuses cytokines, en particulier l’IL-6, responsables d’une augmentation de la synthèse d’hepcidine. L’hepcidine est responsable, via le blocage de l’action de la ferroportine, de l’hypoferrémie et de la diminution de la saturation de la transferrine. D’autres mécanismes indépendants de l’hepcidine expliquent aussi l’augmentation de la ferritine (9).
Nous allons donner quelques exemples d’affections responsables d’anémies inflammatoires avec hyperferritinémie.
L’insuffisance rénale chronique en particulier chez les patients hémodialysés
Il existe souvent une anémie par carence en fer et une anémie inflammatoire (10), l’interprétation du bilan du fer est souvent difficile chez les insuffisants rénaux chroniques en général et chez les dialysés en particulier. L’apport en fer conseillé chez les dialysés n’est pas consensuel. Les données récentes (11) montrent qu’il existe une surcharge en fer (charge hépatique en fer > 50 µmoles/g poids sec) en IRM dans 84 % des cas. La surcharge est sévère (> 200 µmoles/g) dans 30 % des cas. À l’hyperferritinémie d’origine inflammatoire, se surajoute donc l’hyperferritinémie par excès d’apport en fer (Fer IV et transfusions). Des études sont en cours pour préciser les apports nécessaires chez les patients dialysés.
Les hémopathies malignes
Plusieurs études ont montré que les patients atteints d’un myélome multiple et anémiques avaient une augmentation de l’hepcidine urinaire. Il existe aussi une augmentation de l’hepcidine qui est associée à la mortalité chez les patients atteints d’un lymphome non hodgkinien (9).
Les cancers solides
Une méta-analyse récente (12) a montré que plusieurs cancers solides pouvaient être associés à une hyperferritinémie et que la ferritine pouvait servir de biomarqueur en particulier aux stades évolués : tête et cou, poumons, pancréas, reins, foie…
Les maladies de système avec tempête cytokinique
De nombreuses maladies de système ou affections inflammatoires peuvent s’accompagner d’une hyperferritinémie. Parmi ces affections, on peut citer la maladie de Still, le syndrome d’activation macrophagique, le syndrome catastrophique des antiphospholipides et le choc septique. Le diagnostic de syndrome d’activation macrophagique ne peut pas reposer uniquement sur la valeur de la ferritine. Il doit reposer sur le H score qui est validé chez les adultes (13).
Maladie de Gaucher
La maladie de Gaucher est une maladie génétique rare à transmission autosomique récessive, due à un déficit enzymatique en glucocérébrosidase entraînant une accumulation de son substrat, le glucosylcéramide, dans les lysosomes des macrophages. Sa prévalence est estimée à environ 1/60 000 habitants. Il existe trois types de maladie de Gaucher, le plus fréquent est le type 1. Sur le plan clinique, elle associe asthénie, splénomégalie et douleurs osseuses. Sur le plan biologique, on note une thrombopénie, une anémie normocytaire et une hyperferritinémie. Le diagnostic repose sur le dosage de l’activité enzymatique de la glucocérébrosidase dans les leucocytes et sur l’analyse du gène GBA1 (14). L’hyperferritinémie est très fréquente (plus de 80 % des cas) (15). Le coefficient de saturation est le plus souvent inférieur à 45 %. Une surcharge hépatique en fer très modérée serait présente chez un tiers des patients (16).
Hyperthyroïdie
Les études récentes ont inclus des patients avec une maladie de Basedow (17,18) et ont montré que l’hyperferritinémie dépassait rarement 1 000 ng/ ml et que le coefficient de saturation était normal (17). Un tiers des patients avec une maladie de Basedow avait une anémie qui était corrélée négativement à la CRP. L’inflammation et l’augmentation de la synthèse de la ferritine semblent être responsables de l’hyperferritinémie chez les patients atteints d’hyperthyroïdie.
Hyperferritinémie dysmétabolique sans surcharge en fer et hépatosidérose dysmétabolique
Il s’agit probablement de la première cause d’hyperferritinémie en consultation d’hépatologie. Sa fréquence a été estimée à 15 % chez les patients qui avaient un syndrome métabolique selon les critères ATPIII (19). En utilisant les données NANHES III, c’est-à-dire des données en population générale, il a été démontré que l’hyperferritinémie pouvait prédire la survenue d’un diabète (20). Cette augmentation du risque de diabète était observée uniquement chez les patients qui avaient un CST < 45 %. Dans une cohorte coréenne, il a été montré que l’élévation de la ferritine pouvait prédire la survenue d’un syndrome métabolique selon les critères de l’IDF (international diabetes foundation) (21). Ceci veut dire qu’on peut observer une hyperferritinémie avant l’apparition d’un syndrome métabolique complet. Enfin, plusieurs études ont montré qu’il existait un lien entre insulino-résistance et hyperferritinémie dysmétabolique et entre stéatose et hyperferritinémie-dysmétabolique (22). L’hépatosidérose dysmétabolique avec ou sans stéatose a été définie en 1997 par l’équipe de Rennes (23) et associe un ou plusieurs éléments du syndrome métabolique, un coefficient de saturation de la transferrine (CST) < 45 % et une ferritine élevée, supérieure à 1 000 ng/mL chez 10 % des patients (24).
Alcool sans surcharge en fer
La consommation d’alcool est responsable d’une augmentation de la synthèse de la ferritine (25). L’augmentation de la ferritine est observée chez environ la moitié des patients consommateurs excessifs d’alcool (26) et est corrélée à la consommation d’alcool (27). Deux semaines après l’arrêt de l’alcool, la ferritine diminue de 50 % (26).
Syndrome héréditaire hyperferritinémie-cataracte (absence de surcharge en fer)
Le syndrome héréditaire hyperferritinémie-cataracte (SHHC) est une maladie à transmission autosomale dominante. Il associe une hyperferritinémie sans surcharge en fer et une cataracte bilatérale. La fréquence de cette maladie est d’environ 1/200 000 habitants (28). Cette hyperferritinémie est due à une mutation de l’IRE (Iron responsive element) de l’ARNm de la L-ferritine. La ferritine est supérieure à 1 000 ng/ml dans 80 % des cas. La cataracte est présente chez 50 à 100 % des patients.
Maladie de la ferroportine
La ferroportine est la seule molécule connue pour exporter le fer des cellules de l’organisme en particulier au niveau des macrophages, des érythrocytes et des cellules placentaires. Les mutations du gène de la ferroportine, SLC40A1, avec perte de fonction sont responsables d’une surcharge en fer essentiellement macrophagique et donc intéresse à la fois le foie, la rate et la moelle osseuse (29). Cette maladie se transmet selon le mode autosomique dominant, atteint les patients de tout âge de 10 à 80 ans, de toutes ethnies, et est responsable d’une surcharge en fer modérée. La plus grosse série a été rapportée par l’équipe rennaise (30). La ferritine était souvent supérieure à 1 000 ng/mL et le coefficient de saturation dans ¾ des cas était normal ou bas. L’IRM peut être utile au diagnostic quand elle montre une surcharge en fer à la fois hépatique, splénique et osseuse. La fibrose sévère est très rare et intéresse moins de 5 % des patients (30,31). Un dosage de la ferritine chez les parents et les frères et sœurs peut être utile au vu du mode de transmission et de la fréquence de la pénétrance du gène (30,31). Le diagnostic de maladie de la ferroportine doit être affirmé par la biologie moléculaire. En raison de la difficulté à distinguer les surcharges en fer d’origine métabolique de la maladie de la ferroportine, il a été récemment proposé un score dénommé score de la ferroportine (tableau 1) (32). Un score < 9,5 a une très bonne valeur prédictive négative. Le test génétique ne doit être demandé que chez les patients ayant un score > 9.5.
Tableau 1 : Score de la ferroportine| Variable | Points |
|---|
| Sexe Homme Homme | 2.5 0 |
| Age (années) ≤40 41-70 >70 | 5 3 0 |
| HTA ou diabète Oui Non | 3 0 |
| Charge hépatique en fer (µmoles/g) ≤96 97-160 161-200 >200 | 0 1.5 3 4 |
| Ferritine (µg/L) <700 701-1000 1001-1500 >1500 | 0 1.5 2.5 6 |
Valeur maximale : 20.5
Si le score est supérieur à 9,5, il est utile de demander un test génétique
pour rechercher une maladie de la ferroportine
Acéruloplasminémie
L’acéruloplasminémie a été décrite en 1987 par Miyajima et al. L’absence de synthèse d’apocéruloplasmine se manifeste par une surcharge en fer de nombreux organes (les noyaux gris centraux, la substance noire, le noyau rouge, le noyau dentelé du cervelet, la rétine, le foie, le pancréas…). La prévalence a été estimée au Japon à 1/2 000 000 habitants. Elle se transmet sur le mode autosomique récessif (33). Le tableau clinique comporte classiquement trois types de manifestations : des manifestations neurologiques (ataxie cérébelleuse, mouvements anormaux, syndrome extrapyramidal, troubles cognitifs ou démence sous-corticale), un diabète et une rétinite pigmentaire (33). Sur le plan biologique, il existe une anémie microcytaire dans 80 % des cas dans la synthèse de la littérature essentiellement japonaise (33). Les deux études non japonaises (34,35) retrouvent une anémie microcytaire dans 60 % des cas, un coefficient de saturation normal ou bas dans 100 % des cas, une ferritine élevée dans 95 % des cas. La céruléoplasmine est soit indosable soit très basse inférieure à 0,10 g/l dans 85 % des cas. La triade clinique classique doit être remplacée par une triade biologique qui permet un diagnostic et un traitement précoces et associe une anémie microcytaire ou normocytaire, un coefficient de saturation normal ou bas et une hyperferritinémie.
Hémochromatoses
Un groupe d’experts s’est récemment réuni à Heidelberg (36,37) et a proposé la définition suivante de l’hémochromatose : affection génétique responsable d’une hyperabsorption digestive du fer pouvant aboutir progressivement à une surcharge en fer et à des complications pouvant mettre en jeu le pronostic vital des patients (diabète, insuffisance cardiaque, cirrhose et carcinome hépatocellulaire). Le tableau 2 présente la nouvelle classification des hémochromatoses. Les hémochromatoses sont classées en quatre catégories : HFE (HHFE), non HFE (HNONHFE), digénique (HDIGENIQUE), indéterminée (HIND) (36,37). Trois changements importants y figurent : la disparition des chiffres pour nommer chaque type d’hémochromatose, la disparition du terme hémochromatose juvénile, la disparition de la maladie de la ferroportine. La maladie de la ferroportine n’est pas une hémochromatose. En effet le coefficient de saturation est le plus souvent normal, la surcharge en fer est macrophagique et la tolérance aux saignées peut être mauvaise avec apparition d’une anémie. À l’heure actuelle, l’HHFE est découverte à un stade plus précoce et se rencontre essentiellement dans trois situations : dépistage familial chez un patient asymptomatique, anomalies du bilan du fer ou des transaminases chez un patient asymptomatique, patient symptomatique dans 10 % des cas (38). Un test génétique doit être proposé aux parents de premier degré (frèreset sœurs, enfants et parents) des patients atteints d’une HHFE. On peut tester le conjoint quand le couple a au moins deux enfants (39). L’atteinte articulaire et l’élévation des transaminases sont prédictives d’une fibrose sévère (38). Dans les recommandations de l’EASL (37), la fibrose peut être évaluée chez les patients ayant une HHFE par la mesure de l’élasticité hépatique. Cet examen n’a d’intérêt que pour les patients ayant une ferritine > 1 000 ng/mL ou quand il existe une élévation des transaminases (40). Les experts australiens proposent de réaliser des tests biochimiques (APRI et FIB4) (38). Le rôle de C282Y/H63D comme cause d’hémochromatose HFE reste très controversée (Porto 2016). Dans les recommandations américaines, il est conseillé de proposer des saignées aux patients qui ont une ferritine > 1 000 ng/mL et une SHF élevée sans proposer de seuil (41).
Tableau 2 : Principales étiologies des hyperferritinémies selon le coefficient de saturation de la transferrine. Les étiologies avec possible surcharge intra hépatique sont en jaune| Coefficient de saturation < 45% Coefficient de saturation > 45% |
|---|
| Cytolyse (fréquent) | Hépatique | Hémochromatose (peu fréquent) | HFE Non HFE Digénique D’étiologie inconnue |
| Musculaire |
| Inflammation (fréquent) | Infections |
| Maladie de système (PR, LEAD, Still, Vascularite, SAM) | Origine hématologique (peu fréquent) | Hémoglobinopathie (drépanocytose, thalassémie…) |
| Anémie hémolytique |
| Hémopathie maligne /tumeur solide | Syndrome myélodysplasique |
| Insuffisance rénale chronique | Origine hépatique (hors cytolyse) (fréquent) | VHB, VHC, VHD Hépatosidérose dysmétabolique Maladie alcoolique du foie avec surcharge |
| Hyperthyroïdie | Insuffisance hépatique |
| Maladie de Gaucher | Porphyrie cutanée tardive |
| Hyperferritinémie dysmétabolique et hépatosidérose dysmétabolique (fréquent) | Cytolyse aiguë (fréquent) |
| Alcool (fréquent) | |
| Hyperferritinémie-cataracte (très rare) |
| Maladie de la ferroportine (très rare) |
| Acéruloplasminémie (très rare) |
*, LEAD : lupus érythémateux aigu disséminé, PR : polyarthrite rhumatoïde, SAM : Syndrome d’activation Macrophagique
Deux revues de la littérature ont été réalisées pour l’hémochromatose SLC40A1 (31,42). Les mutations de la ferroportine avec GOF (gain of function) sont responsables d’une hémochromatose avec augmentation de l’hepcidine. Elle est exceptionnelle. Elle se manifeste par un coefficient de saturation > 60 %, Une ferritine supérieure à 3 000 ng/mL en moyenne et une fibrose modérée à sévère dans 30 % des cas (31).
Surcharges en fer d’origine hépatique
Alcool
Le coefficient de saturation de la transferrine était augmenté dans environ 15 % des cas (43). La ferritine était augmentée chez 80 % des patients (55/69). Environ un tiers des patients avait une charge hépatique en fer supérieure à la normale (44). À côté de l’augmentation de la synthèse de la ferritine, il existe d’autres mécanismes pour expliquer l’augmentation de la ferritine. Le plus important est la diminution de la synthèse de l’hepcidine par le stress oxydatif induit par l’alcool (45) responsable d’une augmentation de l’absorption digestive du fer.
Hépatosidérose dysmétabolique (HSD) sans ou avec stéatose
Le CST est en réalité élevé dans 20 % des cas environ. La ferritine surestime la surcharge en fer. La stéatose est présente chez la moitié des patients ayant une HSD. Les mécanismes de l’hyperferritinémie ne sont pas encore très clairs. Le rôle de la graisse viscérale, de l’insulino-résistance, de l’augmentation de l’hepcidine et du gène HFE semblent essentiels (24).
Hépatites virales chroniques B et C
Dans une série de 205 patients atteints d’hépatite chronique B, 28 % avaient une augmentation du CST, 20 % avaient une augmentation de la ferritine et 35 % avaient une SHF. Les patients qui avaient une surinfection delta avaient plus souvent une hépatosidérose de grade III-IV (46). D’autres mécanismes ont été proposés pour expliquer l’hyperferritinémie comme l’augmentation de l’hepcidine et la cytolyse (47).
Vingt pour cent des patients atteints d’une hépatite C avaient une augmentation du CST, 20 % avaient une augmentation de la ferritine et un tiers avait une SHF (48). Les facteurs associés de manière indépendante à la SHF étaient la consommation d’alcool et les mutations HFE. Les autres mécanismes proposés sont la baisse de l’hepcidine sérique et la cytolyse (47).
Insuffisance hépatique
Plusieurs équipes ont montré que les patients qui avaient une cirrhose avec insuffisance hépatique avaient dans un tiers des cas un profil du bilan du fer compatible avec le diagnostic d’hémochromatose (CST > 62 % et augmentation de la ferritine). Actuellement, la recherche de la mutation C282Y permet facilement de résoudre les problèmes de diagnostic différentiel sans recourir au rapport charge hépatique en fer/âge (49).
Porphyrie cutanée tardive
Il s’agit de la porphyrie la plus fréquente (50) avec une prévalence estimée à 10/100 000 hab. Elle est due à une inhibition de l’uroporphyrinogène décarboxylase (UROD). Le fer joue un rôle essentiel dans la pathogénie. Les facteurs qui favorisent l’expression de cette maladie sont multiples : le VHC, le VIH, la consommation d’alcool, la consommation de tabac, l’utilisation d’estrogènes (chez les femmes), une mutation de l’UROD et une mutation du gène HFE. Le diagnostic est évoqué chez un adulte de plus de 40 ans ayant une fragilité cutanée puis des lésions bulleuses dans les zones exposées au soleil (dos des mains en particulier). Le coefficient de saturation de la transferrine est augmenté une fois sur 2 et la ferritine peut être normale ou augmentée (51). Le diagnostic repose sur l’augmentation des uroporphyrines dans les urines. Le traitement repose sur les saignées et la chloroquine.
Surcharges en fer secondaires à une maladie hématologique
De nombreuses anémies acquises ou constitutionnelles peuvent être responsables d’une surcharge en fer (tableau 3). Un ou plusieurs mécanismes peuvent expliquer la surcharge en fer observée : l’érythropoïèse inefficace dû à un avortement intramédullaire, l’hémolyse et les transfusions (52,53). L’érythropoïèse inefficace est en cause par exemple dans la surcharge en fer des thalassémies non dépendantes des transfusions. Les principales causes d’anémies acquises avec surcharge en fer sont : les myélodysplasies avec sidéroblastes en couronne (54) et les aplasies médullaires idiopathiques. Les principales causes d’anémies constitutionnelles avec surcharges en fer sont : les thalassémies et la drépanocytose. On détaillera ici uniquement les syndromes myélodysplasiques.
Tableau 3 : Hyperferritinémie et anémie | Saturation < 45% | Saturation > 45% | Surcharge en fer |
|---|
| Anémie | Anémie inflammatoire | Alpha et béta-thalassémies | Non |
| microcytaire | Atransferritinémie | Oui |
| Acéruloplaminémie | Oui |
| Mutation DMT-1 | Oui |
| Oui |
| Myélodysplasie congénitale avec sidéroblastes en couronne (ALAS2, SLC25A38) | Oui |
| Anémie | Hémolyse | Myélodysplasie avec sidéroblastes en couronne (SF3B1) ou myélodysplasie acquise | Non |
| macrocytaire | Carence en B12 | Non |
| Oui |
| Anémies hémolytiques corpusculaires | Oui |
| Anémies hémolytiques auto-immunes | Oui |
| Anémie | Hémolyse | Insuffisance rénale chronique | Non |
| normocytaire | Maladie de Gaucher | Non |
| Oui |
| Drépanocytose | Oui |
| Déficit en pyruvate kinase | Oui |
| CDA* | Oui |
| Aplasie médullaire | Oui |
*congenital dyserythropoietic anemia principalement le type II
Les syndromes myélodysplasiques (SMD) sont dus à une anomalie clonale de la cellule souche hématopoïétique. La prévalence dans la population générale est d’environ 1/100 000 habitants. Sa fréquence est mutipliée par au moins 50 après 70 ans. Les SMD avec sidéroblastes en couronne (54) sont des anémies sidéroblastiques acquises avec une surcharge en fer qui atteignent le plus souvent des sujets âgés de plus de 70 ans. Elles sont responsables d’une anémie macrocytaire arégénérative ou d’une pancytopénie avec une moelle riche en érythroblastes et la présence de plus 15 % de sidéroblastes en couronne. Le diagnostic repose sur la ponction de moelle qui met en évidence la dysplasie et recherche les blastes. La biopsie de moelle évalue la richesse de la moelle, la fibrose et les anomalies cytogénétiques. Une mutation du facteur d’épissage SF3B1 est retrouvée dans 80 % des anémies sidéroblastiques acquises. Les anémies sidéroblastiques congénitales avec surcharge en fer sont très rares (55).
Évaluation de la charge hépatique en fer
La charge hépatique en fer peut être évaluée grâce à une biopsie hépatique ou une IRM. En raison du caractère invasif de la ponction biopsie hépatique, on utilise beaucoup plus souvent la quantification du fer par IRM. Trois méthodes différentes sont disponibles : la première utilise une séquence en écho de gradient et compare l’intensité du signal du foie à celle des muscles para-vertébraux, la deuxième utilise la relaxométrie avec une séquence en écho de spin (T2 et R2 = 1 000/T2), la troisième utilise la relaxométrie en écho de gradient (T2* et R2* = 1 000/T2*) (56,57). En 2017, Gandon et al. ont proposé d’utiliser le R2* pour quantifier la CHF (charge hépatique en fer) et de le coupler au calcul de la graisse hépatique par mesure de la densité du proton (MRQuantif). Là encore, il existe un logiciel gratuit disponible sur le site de l’université de Rennes pour calculer la CHF (56). L’importance de la CHF peut être classée en cinq catégories : 1 à 2N (N = 36 µmoles/g), 2 à 3N, 3 à 4N, 4 à 8N et > 8N (56). On considère qu’une SHF est significative lorsqu’elle est > 100 µmol/g où supérieure à 3N. L’IRM permet le suivi sous traitement. Il a été montré que la charge hépatique en fer était corrélée au stock en fer de l’organisme (58). La CHF est donc utilisée pour suivre l’efficacité d’un traitement chélateur, de même que le T2* cardiaque en particulier dans les surcharges en fer de la thalassémie dépendante des transfusions (59). Les indications de l’IRM sont assez bien définies pour les surcharges en fer hématologiques. Les indications de l’IRM des surcharges non hématologiques ne sont pas encore très clairement définies. Il semblerait qu’une valeur de la ferritine supérieure à 500 ng/mL serait une bonne indication d’IRM dans les NAFLD. Il nous semble qu’une valeur de ferritine supérieure à 1 000 ng/mL pourrait également être discutée chez les patients ayant un coefficient de saturation de la transferrine < 45 % comme le suggèrent les recommandations anglaises (60).
Conduite à tenir devant une hyperferritinémie
L’hyperferritinémie est une cause extrêmement fréquente de consultation en hématologie, médecine interne et hépato-gastroentérologie. Une bonne connaissance du contexte clinique, des antécédents familiaux et personnels du patient, des toxiques consommés, des étiologies des hyperferritinémies (tableau 2) et un bilan de débrouillage (tableau 4) permettent dans 95 % des cas d’arriver à un diagnostic précis.
Tableau 4 : Bilan étiologique de première intention d’une hyper-ferritinémie| INTERROGATOIRE |
|---|
| Antécédents familiaux | Hyperferritinémie, cataracte, saignée |
| Antécédents personnels | Chirurgie de la cataracte jeune |
| Syndrome métabolique |
| Historique du poids : actuel et maximal, taille, IMC |
| Consommation d’alcool actuelle ou passée : AUDIT-C |
| Maladie hépatique (alcool, virus, NAFLD) |
| Insuffisance rénale avancée |
| Maladie hématologique, transfusions multiples |
| Activité physique | Exercice sportif intense (musculation, course de fond…) |
| Signes fonctionnels | Arthralgie, troubles de la libido, aménorrhée, |
| Traitement | Fer oral, IV, compléments alimentaires |
| EXAMEN CLINIQUE |
|---|
| Poids taille IMC tour de taille |
| Mélanodermie, hypogonadisme |
| Signe d’insuffisance hépatocellulaire ou d’hypertension portale |
| Signe d’hémopathie/ néoplasie |
EXAMENS DE PREMIÈRE INTENTION
Coefficient de saturation de la transferrine répété à deux reprises |
|---|
| CST < 45% | CST > 45% |
| NFS, réticulocytes | NFS plaquettes |
| TSH TP, ASAT ALAT GGT PAL, bilirubine, FIB4 Glycémie, insulinémie à jeun, EAL, créatinine, EPP Sérologies VHC, VHB, VIH Sérologies VHC, VHB, VIH CRP (syndrome inflammatoire) Haptoglobine, LDH (hémolyse) CPK (myolyse) Echographie hépatique Elastographie hépatique avec éventuellement CAP | Mutation HFE C282Y TP, ASAT ALAT GGT PAL, bilirubine, FIB4 Glycémie, insulinémie à jeun, EAL, créatinine, EPP Sérologies VHC, VHB, VIH Haptoglobine, LDH (hémolyse) Echographie hépatique Elastographie hépatique avec éventuellement CAP |
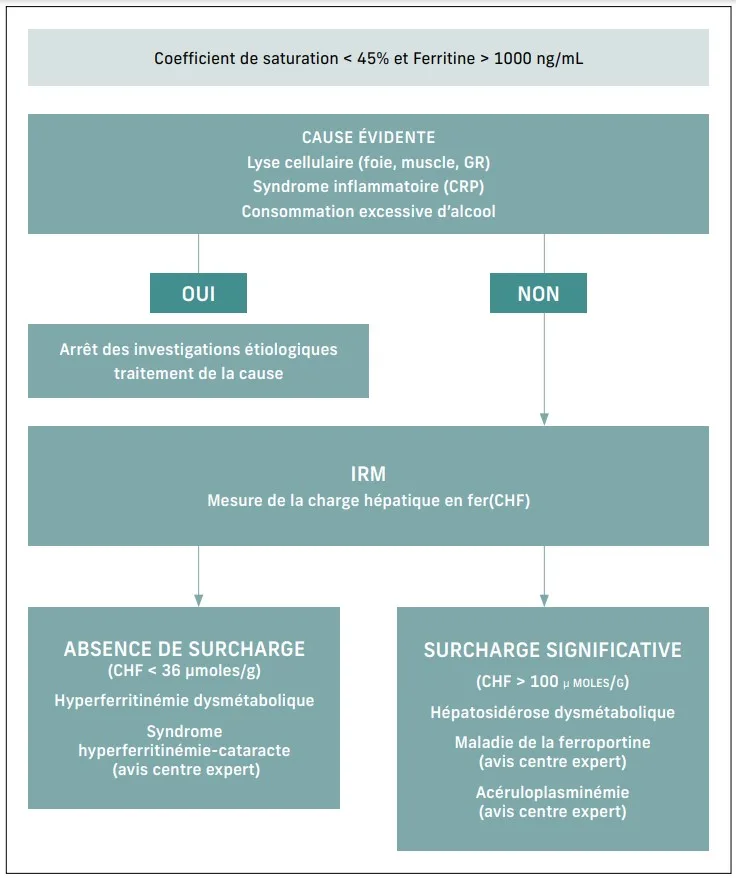
Trois paramètres sont fondamentaux dans cette démarche : la valeur du coefficient de saturation (inférieure ou supérieure à 45 %), de la ferritine (inférieure ou supérieure à 1 000 ng/mL) et la fréquence de chaque pathologie. Dans le tableau 2, la valeur du coefficient de saturation de chaque étiologie est précisée ainsi que la fréquence estimée des différentes étiologies vues en consultation d’hépatologie. Les données en population générale sont inexistantes. Il est classique de dire que les causes les plus fréquentes d’hyperferritinémie en milieu hépatologique sont l’alcool, l’inflammation, l’hyperferritinémie dysmétabolique (hyperF dysmétabolique), la cytolyse et les hépatopathies chroniques. Il est probable que l’hyperF dysmétabolique soit la première cause en milieu hépatologique. Quand le coefficient de saturation est normal et la ferritine est inférieure à 1 000 ng/mL, il faut proposer un arrêt de la consommation d’alcool, une prise en charge par une diététicienne et encourager le patient à pratiquer une activité physique régulière. Si la ferritine est supérieure à 1 000 ng/mL, une IRM pour mesure de la charge hépatique en fer (60) et de la graisse hépatique est nécessaire (MRQuantif). Si l’IRM montre une CHF normale, il faut rechercher une mutation du FTL (FerriTin Light chain). S’il existe une SHF, il faudra rechercher une maladie de la ferroportine (calcul du score de la ferroportine, recherche à l’IRM d’une surcharge en fer de la rate et des vertèbres) et une acéruloplasminémie (dosage de la céruloplasmine). L’avis du réseau du CRFer est indispensable dans les cas difficiles (figure 1).

Figure 1 : Conduite à tenir devant un coefficient de saturation
de la transferrine < 45 % et une hyperferritinémie
Quand on suspecte une hémochromatose (cs > 45 % à 2 reprises et ferritine augmentée), il faut demander une recherche de la mutation C282Y. Si le patient est C282Y homozygote, le patient a une hémochromatose HFE. La recherche d’une hémochromatose non-HFE nécessite la recherche d’un panel de gènes. L’aide du réseau CNfer est indispensable.
Pour les surcharges en fer d’origine hématologique, les signes d’appel les plus importants sont : l’anémie et la nécessité de transfuser régulièrement. Il faut évoquer en premier la myélodysplasie acquise, l’anémie est le plus souvent macrocytaire arégénérative (figure 2).

Figure 2 : Conduite à tenir devant un coefficient de saturation de la transferrine > 45 % et une hyperferritinémie
Traitement des surcharges en fer
L’objectif du traitement est de prévenir les complications liées aux surcharges d’organes et améliorer la qualité de vie. La diminution des apports en fer pour prévenir ou traiter la surcharge n’est pas recommandée par l’EASL (37). Les saignées restent le traitement de référence des surcharges en fer non hématologiques hors anémie, notamment pour l’hémochromatose (37). À ce jour aucune étude de qualité suffisante n’a montré l’intérêt du traitement par saignées des patients atteints d’une hépatopathie chronique et d’hépatosidérose. Pour l’hémochromatose, l’objectif est un taux de ferritinémie normale basse à 50-100 µg/L sans anémie avec une bonne tolérance clinique. Le traitement médicamenteux chélateur est indiqué en cas de contre-indication aux saignées en cas d’hémochromatose, d’anémies avec surcharge en fer ou en prophylaxie primaire de la surcharge en fer post-transfusionnelle en cas de maladie hématologique. Sa tolérance est cependant moyenne. Trois molécules sont actuellement disponibles : Deferoxamine (DFO), Deferasirox (DFX) et Deferiprone (DFP) (61-64). Les deux objectifs du traitement chélateur sont de faire baisser rapidement le fer non lié à la transferrine, dont la toxicité a été soulignée par de multiples études, et de réduire progressivement les stocks de fer tissulaire en particulier hépatique et cardiaque. Le tableau 5 résume les principales indications, modalités d’administration et les principaux effets secondaires de ces trois produits.
Tableau 5 : Propriétés des trois chélateurs du fer | Deferoxamine | Deferiprone | Deferasirox |
|---|
| AMM pour la surcharge en fer | 1997 | 1999 | 2006 |
| Nom commercial | Desferal® | Ferriprox® | Exjade® |
| Demi-vie | 20 min | 2 à 3 h | 8-16h |
| Dose recommandée | 30 à 60 mg/kg/j | 75-100 mg/kg/j en 3 prises | 14-28 mg/kg/j en 1 prise |
| Voie d’administration | Sous cutanée ou IV | Per os (comprimés ou liquide) | Per os (comprimés pelliculés) |
| Voie d’élimination | Urinaire (50%) et fécale (50%) | Urinaire 90% | Fécale 85% |
| Indications | - Hémosidérose secondaire
- Hémochromatose non curable par saignées.
| - Thalassémie dépendante des transfusions (TDT) lorsque le traitement chélateur en cours est contre-indiqué ou inadapté
- TDT avec atteinte cardiaque
| - TDT
- Traitement de la surcharge en fer chronique secondaire à des transfusions sanguines lorsque le traitement par la déféroxamine est contre-indiqué ou inadapté
- Thalassémie non dépendante des transfusions
|
| Principales contre-indications | - Insuffisance rénale sévère non dialysée
- Infection bactérienne non traitée
- Grossesse
| | - Clairance de la créatinine < 60 ml/min
- Grossesse
|
| Effet sur la SHF | +++ | +++ | +++ |
| Effet sur SCF | + | +++ | ++ |
| Effet sur la SPF | + | + | + |
| Effet sur la SEF | + | ++ | + |
| Principaux effets secondaires dont la fréquence > 5% | - Retard de croissance
- Réaction au point de ponction
- Allergie
| - Troubles digestifs
- Douleurs articulaires
- Augmentation des transaminases
- Neutropénie
- Couleur brunâtre des urines
| - Troubles digestifs
- Rash
- Cytolyse
- Augmentation de la créatinine
|
| Effet secondaire rare mais grave ou justifiant d’une surveillance spécifique | Ototoxicité Toxicité rétinienne | - Agranulocytose (0.8 % chez les hommes et 2.4 % chez les femmes)
- Déficit en zinc chez les diabétiques
| - Insuffisance rénale
- Tubulopathie de type Fanconi
- Perte d’audition
- Cataracte
|
| Coût annuel en euros | 6 300 | 6 200 | 26 000 |
| Fréquence des arrêts pour effet secondaire | 4 % | 5 % | 6 % |
*SHF : surcharge hépatique en fer, SCF : surcharge cardiaque en fer, SPF : surcharge pancréatique en fer, SEF : surcharge endocrinienne en fer en particulier hypophysaire.
Conclusion
L’hyperferritinémie est une des principales causes de consultation en hépatologie. Un bilan étiologique de débrouillage avec le coefficient de saturation de la transferrine permet d’identifier facilement les principales causes, dominées par l’hyperferritinémie liée au syndrome métabolique. Lorsque le coefficient de saturation est supérieur à 45 % une hémochromatose HFE doit être suspectée et une mutation homozygote C282Y doit être recherchée. Lorsque le diagnostic est incertain et la ferritine supérieure à 1 000 µg/l, l’IRM est l’examen de référence permettant d’affirmer l’existence d’une surcharge en fer et/ou en graisse. Les autres causes d’hyperferritinémie doivent être connues afin de ne pas méconnaitre un diagnostic comme la maladie de la ferroportine dont le diagnostic devient désormais plus facile avec l’utilisation du score de la ferroportine. En cas d’anémie, une cause hématologique doit être recherchée. La phlébotomie thérapeutique reste le traitement de référence de l’hémochromatose HFE avec l’objectif de maintenir une ferritinémie entre 50 et 100 µg/L. Ce traitement n’est cependant pas recommandé en cas d’hépatosidérose métabolique.
Bibliographie
- Sandnes M, Ulvik RJ, Vorland M, et al. Hyperferritinemia-A Clinical Overview. J Clin Med. 2021;10:2008.
- Beaumont C, Karim Z. [Iron metabolism: State of the art]. Rev Med Interne. 2013;34:17-25.
- Jenkitkasemwong S, Wang CY, Coffey R, Zhang W, Chan A, Biel T, et al. D. SLC39A14 Is Required for the Development of Hepatocellular Iron Overload in Murine Models of Hereditary Hemochromatosis. Cell Metab. 2015;22:138-50.
- Cabantchik ZI, Breuer W, Zanninelli G, Cianciulli P. LPI-labile plasma iron in iron overload. Best Pract Res Clin Haematol. 2005;18:277-87.
- Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46:678-84.
- Nemeth E, Ganz T. Hepcidin and Iron in Health and Disease. Annu Rev Med. 2023;74:261-77.
- Donker AE, Raymakers RA, Vlasveld LT, van Barneveld T, Terink R, Dors N, et al. Practice guidelines for the diagnosis and management of microcytic anemias due to genetic disorders of iron metabolism or heme synthesis. Blood. 2014;123:3873-86
- Lorcerie B, Audia S, Samson M, et al. Cytolyse sans surcharge en fer. Diagnosis of hyperferritinemia in routine clinical practice. Presse Med. 2017;46:e329-38.
- Weiss G, Ganz T, Goodnough LT. Anemia of inflammation. Blood. 2019;133:40-50.
- Majoni SW, Lawton PD, Rathnayake G, et al. Narrative Review of Hyperferritinemia, Iron Deficiency, and the Challenges of Managing Anemia in Aboriginal and Torres Strait Islander Australians With CKD. Kidney Int Rep. 2020;6:501-12.
- Rostoker G, Griuncelli M, Loridon C, et al. Hemodialysis-associated hemosiderosis in the era of erythropoiesis-stimulating agents: a MRI study. Am J Med. 2012;125:991-9.e1.
- Ramírez-Carmona W, Díaz-Fabregat B, Yuri Yoshigae A, et al. Are Serum Ferritin Levels a Reliable Cancer Biomarker? A Systematic Review and Meta-Analysis. Nutr Cancer. 2022;74:1917-26.
- Ruscitti P, Di Cola I, Di Muzio C, et al. Expanding the spectrum of the hyperferritinemic syndrome, from pathogenic mechanisms to clinical observations, and therapeutic implications. Autoimmun Rev. 2022;21:103114.
- Nguyen Y, Stirnemann J, Belmatoug N. Gaucher disease: A review. Rev Med Interne. 2019;40:313-22.
- Lorenz F, Pawłowicz E, Klimkowska M, et al. Ferritinemia and serum inflammatory cytokines in Swedish adults with Gaucher disease type 1. Blood Cells Mol Dis. 2018;68:35-42.
- Bohte AE, van Dussen L, Akkerman EM, et al. Liver fibrosis in type I Gaucher disease: magnetic resonance imaging, transient elastography and parameters of iron storage. PLoS One. 2013;8:e57507.
- Fischli S, von Wyl V, Trummler M, et al. Iron metabolism in patients with Graves’ hyperthyroidism. Clin Endocrinol (Oxf). 2017;87:609-16.
- Krygier A, Szczepanek-Parulska E, Filipowicz D, et al. Changes in serum hepcidin according to thyrometabolic status in patients with Graves’ disease Endocr Connect. 2020;9:234-42.
- Bozzini C, Girelli D, Olivieri O, et al. Prevalence of body iron excess in the metabolic syndrome. Diabetes Care. 2005;28:2061-3.
- Ford ES, Cogswell ME. Diabetes and serum ferritin concentration among U.S. adults. Diabetes Care. 1999;22:1978-83.
- Park SK, Ryoo JH, Kim MG, Shin JY. Association of serum ferritin and the development of metabolic syndrome in middle-aged Korean men: a 5-year follow-up study. Diabetes Care. 2012;35:2521-6.
- Brudevold R, Hole T, Hammerstrøm J. Hyperferritinemia is associated with insulin resistance and fatty liver in patients without iron overload. PLoS One. 2008;3:e3547.
- Moirand R, Mortaji AM, Loréal O, et al. A new syndrome of liver iron overload with normal transferrin saturation. Lancet. 1997;349:95-7.
- Rametta R, Fracanzani AL, Fargion S, et al. Dysmetabolic Hyperferritinemia and Dysmetabolic Iron Overload Syndrome (DIOS): Two Related Conditions or Different Entities? Curr Pharm Des. 2020;26:1025-35.
- Moirand R, Kerdavid F, Loréal O, et al. Regulation of ferritin expression by alcohol in a human hepatoblastoma cell line and in rat hepatocyte cultures. J Hepatol. 1995 Oct;23:431-9.
- Bell H, Skinningsrud A, Raknerud N, Try K. Serum ferritin and transferrin saturation in patients with chronic alcoholic and non-alcoholic liver diseases. J Intern Med. 1994 Sep;236(3):315-22.
- Leggett BA, Brown NN, Bryant SJ, et al. Factors affecting the concentrations of ferritin in serum in a healthy Australian population. Clin Chem. 1990;36:1350-5.
- Craig JE, Clark JB, McLeod JL, et al. Hereditary hyperferritinemia-cataract syndrome: prevalence, lens morphology, spectrum of mutations, and clinical presentations. Arch Ophthalmol. 2003;121:1753-61.
- Rishi G, Subramaniam VN. Biology of the iron efflux transporter, ferroportin. Adv Protein Chem Struct Biol. 2021;123:1-16.
- Le Lan C, Mosser A, Ropert M, et al. Sex and acquired cofactors determine phenotypes of ferroportin disease. Gastroenterology. 2011;140:1199-1207.e1-2.
- Mayr R, Janecke AR, Schranz M, et al. Ferroportin disease: a systematic meta-analysis of clinical and molecular findings. J Hepatol. 2010;53:941-9.
- Landemaine A, Hamdi-Roze H, Cunat S, et al. A simple clinical score to promote and enhance ferroportin disease screening. J Hepatol. 2022;76:568-76.
- Miyajima H, Hosoi Y. Aceruloplasminemia. 2003 Aug 12 [updated 2018 Sep 27]. In: Adam MP, Everman DB, Mirzaa GM, et al. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2022.
- Pelucchi S, Mariani R, Ravasi G, et al. Phenotypic heterogeneity in seven Italian cases of aceruloplasminemia. Parkinsonism Relat Disord. 2018;51:36-42.
- Vila Cuenca M, Marchi G, Barqué A, et al. Genetic and Clinical Heterogeneity in Thirteen New Cases with Aceruloplasminemia. Atypical Anemia as a Clue for an Early Diagnosis. Int J Mol Sci. 2020;21:2374.
- Girelli D, Busti F, Brissot P, et al. Hemochromatosis classification: update and recommendations by the BIOIRON Society. Blood. 2022;139:3018-29.
- EASL Clinical Practice Guidelines on haemochromatosis. J Hepatol. 2022;77:479-502.
- Olynyk JK, Ramm GA. Hemochromatosis. N Engl J Med. 2022;387:2159-70.
- Porto G, Brissot P, Swinkels DW, et al. EMQN best practice guidelines for the molecular genetic diagnosis of hereditary hemochromatosis (HH). Eur J Hum Genet. 2016;24:479-95.
- Fitzsimons EJ, Cullis JO, Thomas DW, et al. British Society for Haematology. Diagnosis and therapy of genetic haemochromatosis (review and 2017 update). Br J Haematol. 2018;181:293-303.
- Kowdley KV, Brown KE, Ahn J, Sundaram V. ACG Clinical Guideline: Hereditary Hemochromatosis. Am J Gastroenterol. 2019;114:1202-18.
- Zhang W, Lv T, Huang J, Ou X. Type 4B hereditary hemochromatosis associated with a novel mutation in the SLC40A1 gene: A case report and a review of the literature. Medicine 2017;96:e8064.
- Bell H, Skinningsrud A, Raknerud N, Try K. Serum ferritin and transferrin saturation in patients with chronic alcoholic and non-alcoholic liver diseases. J Intern Med. 1994;236:315-22.
- Chapman RW, Morgan MY, Laulicht M, et al. Hepatic iron stores and markers of iron overload in alcoholics and patients with idiopathic hemochromatosis. Dig Dis Sci. 1982;27:909-16.
- Harrison-Findik DD, Schafer D, Klein E, et al. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J Biol Chem. 2006;281:22974-82.
- Sebastiani G, Tempesta D, Alberti A. Hepatic iron overload is common in chronic hepatitis B and is more severe in patients coinfected with hepatitis D virus. J Viral Hepat. 2012;19:e170-6.
- Czaja AJ. Review article: iron disturbances in chronic liver diseases other than haemochromatosis – pathogenic, prognostic, and therapeutic implications. Aliment Pharmacol Ther. 2019;49:681-701.
- Valenti L, Pulixi EA, Arosio P, et al. Relative contribution of iron genes, dysmetabolism and hepatitis C virus (HCV) in the pathogenesis of altered iron regulation in HCV chronic hepatitis. Haematologica. 2007;92:1037-42.
- Cotler SJ, Bronner MP, Press RD, et al. End-stage liver disease without hemochromatosis associated with elevated hepatic iron index. J Hepatol. 1998;29:257-62.
- Bissell DM, Anderson KE, Bonkovsky HL. Porphyria. N Engl J Med. 2017;377:862-72.
- Skowron F, Bérard F, Grézard P, Wolf F, Morel Y, Perrot H. [Role of the hemochromatosis gene in prophyria cutanea tarda. Prospective study of 56 cases]. Ann Dermatol Venereol. 2001;128:600-4.
- Allali S, de Montalembert M, Brousse V, et al. Management of iron overload in hemoglobinopathies. Transfus Clin Biol. 2017;24:223-6.
- Cazzola M. Ineffective erythropoiesis and its treatment. Blood. 2022;139:2460-70.
- Cazzola M. Myelodysplastic Syndromes. N Engl J Med. 2020;383:1358-74.
- Abu-Zeinah G, DeSancho MT. Understanding Sideroblastic Anemia: An Overview of Genetics, Epidemiology, Pathophysiology and Current Therapeutic Options. J Blood Med. 2020;11:305-18.
- Henninger B, Alustiza J, Garbowski M, Gandon Y. Practical guide to quantification of hepatic iron with MRI. Eur Radiol. 2020;30:383-93.
- Labranche R, Gilbert G, Cerny M, et al. Liver Iron Quantification with MR Imaging: A Primer for Radiologists. Radiographics. 2018;38:392- 412.
- Paisant A, d’Assignies G, Bannier E, et al. MRI for the measurement of liver iron content, and for the diagnosis and follow-up of iron overload disorders. Presse Med. 2017;46:e279-e287.
- Angelucci E, Brittenham GM, McLaren CE, et al. Hepatic iron concentration and total body iron stores in thalassemia major. N Engl J Med. 2000;343:327-31.
- Cullis JO, Fitzsimons EJ, Griffiths WJ, Tsochatzis E, Thomas DW; British Society for Haematology. Investigation and management of a raised serum ferritin. Br J Haematol. 2018;181:331-40.
- Hider RC, Hoffbrand AV. The Role of Deferiprone in Iron Chelation. N Engl J Med. 2018;379:2140-50.
- Pinto VM, Forni GL. Management of Iron Overload in Beta-Thalassemia Patients: Clinical Practice Update Based on Case Series. Int J Mol Sci. 2020;21:8771.
- Palumbo GA, Galimberti S, Barcellini W, et al. From Biology to Clinical Practice: Iron Chelation Therapy With Deferasirox. Front Oncol. 2021;11:752192.
- Shah FT, Porter JB, Sadasivam N, et al. Guidelines for the monitoring and management of iron overload in patients with haemoglobinopathies and rare anaemias. Br J Haematol. 2022;196:336-50.
Abréviations
CHF : charge hépatique en fer
ERFE : érythroferrone
FTL : FerriTin Light chain
IDF : international diabetes foundation
NTBI : non transferrin bound iron
SHF : surcharge hépatique en fer