Objectifs pédagogiques
- Savoir évoquer une maladie de Wilson
- Connaître la démarche diagnostique
- Connaître les critères pronostiques
- Connaître les modalités du traitement et de sa surveillance
- Savoir mener l’enquête génétique
Toute reproduction ou réécriture, totale ou partielle, sans l’accord préalable écrit de la FMC HGE est interdite.
Orphalan ; Alexion
Maladie de Wilson ; Diagnostic ; Traitement
La maladie de Wilson (MW) est une maladie génétique rare due à l’accumulation excessive de cuivre dans l’organisme, principalement dans le foie et dans le cerveau. Il s’agit d’une affection monogénique de transmission autosomique récessive, due à la présence de mutations dans le gène localisé sur le chromosome 13 codant pour une protéine transporteuse du cuivre essentiellement localisée dans le foie, l’ATPase 7B. En cas de dysfonctionnement de l’ATPase 7B, il existe un défaut d’excrétion biliaire du cuivre qui a pour conséquence une accumulation toxique de cuivre libre, initialement dans le foie puis secondairement dans les autres organes via le passage dans la circulation générale (1).
La prévalence génétique classique de la MW est de 1/30 000 mais serait peut-être plus fréquente d’après certaines études génétiques récentes. La prévalence des hétérozygotes varie entre 1/40 et 1/90. La prévalence clinique de la MW est certainement sous-estimée. Selon les données médico- administratives nationale SNIIR-AM (2013), elle serait de l’ordre de 1,5/100 000 en France. La différence entre prévalence clinique et génétique pourrait être expliquée par une pénétrance incomplète de la maladie et/ ou la présence de gènes modificateurs (non connus) à l’origine de phénotypes peu sévères et/ ou une maladie encore largement sous diagnostiquée (2).
Le diagnostic de la MW repose sur un faisceau d’arguments cliniques, biologiques et morphologiques (tableau 1). Le score de Leipzig est un score qui a été proposé par des experts internationaux permettant d’aider le clinicien à établir un diagnostic de MW à partir de ces différents items mais n’a pas été réactualisé depuis 2003, ne prenant pas en compte les nouveaux biomarqueurs actuellement disponibles (3) (tableau 2).
| Forme présymptomatique (dépistage) | Forme hépatique chronique asymptomatique (dépistage) | Forme hépatique chronique symptomatique | Forme neurologique | Forme hépatique fulminante | |
|---|---|---|---|---|---|
| Manifestations | BH normal Absence de symptômes neurologiques | Cytolyse Absence de symptômes neurologiques | Cytolyse + asthénie +/- douleur abdo +/- signes neurologiques | Signes neuro au premier plan +/- signes hépatiques associés | Ictère, douleur abdo, anémie hémolytique +/- symptômes neurologiques |
| Cuivre sérique total | diminué | diminué | diminué | diminué | augmenté |
| Céruloplasmine (0,20-0,40 g/L) | diminuée | diminuée | diminuée | diminuée | diminuée ou normale |
| Cuivre échangeable | normal | normal | normal ou augmenté | augmenté | très augmenté |
| REC | >15% | >15% | >18,5% | >18,5% | ? |
| Cuprurie/24h | peut être normale | modérément augmentée (40 à 100 µg/j) (0,6-1,6 µmol/j) | en général augmentée <ou> 100 µg/j (<ou> 1,6 µmol/j) | Augmentée >100µg/j (>1,6 µmol/j) | Très augmentée >100 µg/j voire >1000 µg/j (>1,6 µmol/j voire >16 µmol/j) |
| Dosage pondéral Cuivre hépatique | ? | ? | augmenté (>250 µg/g) (>4 µmol/g) | augmenté (>250 µg/g) (>4 µmol/g) | augmenté (>250 µg/g) (>4 µmol/g) |
| Anneau de KF | absent | le plus souvent absent | environ 50% | >95% | environ 50% |
| Anomalie à l’IRM cérébrale | normal | le plus souvent normale | environ 50% | >95% | environ 50% |
| Biologie moléculaire | 2 mutations ATPase 7B | 2 mutations ATPase 7B | 2 mutations ATPase 7B | 2 mutations ATPase 7B | 2 mutations ATPase 7B |
| Paramètres | Points |
|---|---|
| CARACTÉRISTIQUES CLINIQUES SPÉCIFIQUES | |
| Anneau de Kayser-Fleischer (examen à la lampe à fente) | |
| Présence | 2 |
| Absence | 0 |
| Symptômes neuropsychiatriques évocateurs de maladie de Wilson (ou lésions radiologiques typiques sur l’IRM cérébrale) | |
| Présence | 2 |
| Absence | 0 |
| Anémie hémolytique à test de Coombs négatif (+ élévation du cuivre sérique) | |
| Présence | 1 |
| Absence | 0 |
| TESTS BIOLOGIQUES | |
| Cuprurie /24h (en absence d’hépatite aigue) | |
| Valeur normale (<100 µg/24h chez l’adulte et <40 µg/24h chez l’enfant) | 0 |
| 1-2x la valeur normale | 1 |
| >2x la valeur normale | 2 |
| Valeur basale normale mais > 500 µg/24h après test à la D-penicillamine (2x500mg) | 2 |
| Dosage du cuivre intra-hépatique | |
| Valeur normale (< 50 µg/g de poids sec) | -1 |
| jusqu’à 5x la valeur normale | 1 |
| >5x la valeur normale | 2 |
| Coloration positive des hépatocytes par la Rhodanine (seulement si le dosage du cuivre intrahépatique n’est pas disponible) | |
| Présence | 0 |
| Absence | 1 |
| Céruloplasmine sérique (dosage par méthode néphélométrique, valeur normale : > 20 mg/dL) | |
| Valeur normale | 0 |
| 10-20 | 1 |
| <10 | 2 |
| Recherche de mutation du gène ATP7B par biologie moléculaire | |
| Mutations pathogènes sur les 2 chromosomes | 4 |
| Mutations pathogènes sur un seul chromosome | 1 |
| Absence de mutations pathogènes détectées | 0 |
| SCORE TOTAL (si des données ne sont pas disponibles pour un item mettre un score 0) | |
| Evaluation du score diagnostique pour la maladie de Wilson | |
| 4 points ou plus : Diagnostic de maladie de Wilson hautement probable | |
| 2-3 : Diagnostic de maladie de Wilson possible mais non certain, compléter par des explorations complémentaires | |
| 0-1 point : Diagnostic de maladie de Wilson peu probable | |
La MW est un caméléon clinique auquel il faut savoir penser. Elle touche habituellement le sujet jeune, classiquement entre 5 ans et 40 ans, avec un pic de révélation lors des 2e et 3e décennies, toutefois des formes symptomatiques de révélations précoces (2 ans) ou tardives (>70 ans) ont été décrites. Dans ces conditions, l’âge en soi ne permet pas d’éliminer un diagnostic de MV.
La MW peut se présenter par un grand nombre de manifestations principalement hépatiques, neurologiques et/ ou psychiatriques avec de multiples combinaisons possibles. L’atteinte hépatique est variable et peut se manifester par de simples anomalies des tests hépatiques, une stéatose hépatique et/ ou une hépatomégalie en imagerie, une hépatite aiguë ou chronique, une cirrhose ou une hépatite fulminante. L’atteinte neurologique peut accompagner l’atteinte hépatique ou être au premier plan. La dysarthrie est le symptôme le plus fréquemment observé lors du diagnostic pouvant être associé à une atteinte dystonique, un tremblement, des troubles de la marche, un syndrome parkinsonien et des mouvements anormaux choréiques. La dysarthrie, la dystonie, le tremblement et le syndrome parkinsonien peuvent également être des symptômes isolés lors de la révélation de la MW. Les manifestations psychiatriques sont probablement sous-estimées et sont également variées : troubles de l’humeur incluant la dépression et des troubles bipolaires, troubles de la personnalité incluant comportement antisocial et désinhibition sexuelle, troubles cognitifs. Tout patient porteur d’une MW doit faire l’objet d’un examen neurologique approfondi et en cas d’atteinte neurologique avérée, l’évaluation neurologique devra être faite au moyen d’une échelle dédiée, appelée UWDRS (Unified Wilson Disease Rating Scale) (4), qui a un intérêt pronostique mais également permettra un suivi de l’évolution de l’atteinte sous traitement.
En cas de suspicion d’une MW, les tests cupriques importants à réaliser sont les dosages du cuivre échangeable, de la céruloplasmine, de la cuprémie totale et de la cuprurie des 24 heures. Des résultats anormaux permettront d’instaurer un traitement chélateur sans délai en attendant la confirmation définitive du diagnostic par l’étude en biologie moléculaire du gène ATP7B (tableau 1) (5).
En dehors de l’atteinte fulminante, le diagnostic biologique repose sur des anomalies du bilan cuprique avec la triade classique « hypocuprémie- hypocéruloplasminémie-hypercuprurie » associée à une élévation du REC (ratio cuivre échangeable/cuivre total).
Bien qu’étant une maladie de surcharge de l’organisme en cuivre, on observe habituellement, en dehors des atteintes aiguës sévères, une diminution du cuivre sérique total chez le patient atteint de MW qui s’explique par la forte diminution de la quantité de cuivre lié à la céruléoplasmine qui représente la fraction majoritaire chez le sujet sain (> 90 %). La quantité de cuivre toxique, non lié à la céruloplasmine, est le plus souvent augmenté chez le sujet atteint de la maladie de Wilson. Le dosage de la fraction de cuivre non lié à la céruloplasmine, appelé cuivre échangeable, est un des éléments importants du diagnostic : le REC (ratio cuivre échangeable/cuivre sérique total) est le plus souvent augmenté (> 18,5 %) chez les patients atteints de MW alors qu’il est habituellement bas (< 10 %) chez les sujets indemnes notamment les hétérozygotes ou les patients atteints d’une autre hépatopathie chronique. Un taux augmenté de cuivre échangeable > 2 µmol/L est très souvent rencontré chez les patients atteints de MW en cas d’atteinte extra-hépatique (6,7). La figure 1A présente la liste des laboratoires réalisant le dosage du cuivre échangeable.
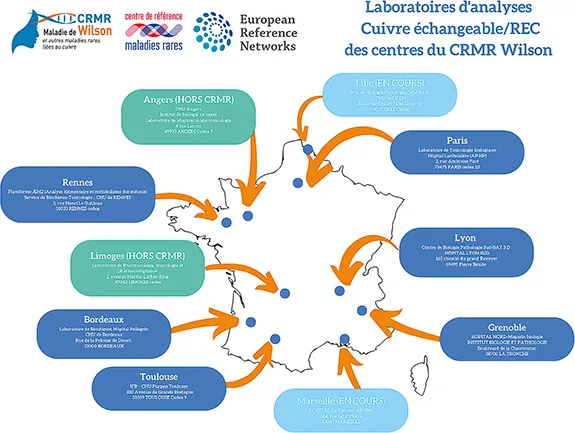
Figure 1A : Liste des laboratoires réalisant le dosage du cuivre échangeable
(mise à jour décembre 2022)
Une diminution importante de la céruloplasminémie (< 0,10 g/L) est très évocateur du diagnostic de MW. Des taux intermédiaires (0,10- 0,20 g/L) doivent faire rechercher une maladie de Wilson mais sont moins spécifiques et des taux normaux (> 0,20 g/L) peuvent être observés chez 5 à 20 % des patients, un taux normal n’éliminant donc pas formellement le diagnostic de MW. La céruloplasminémie peut être diminuée dans d’autres situations que la MW notamment en cas de cirrhose en lien avec l’insuffisance hépatique, mais également en cas de malabsorption, de dénutrition, de syndrome néphrotique, de carence acquise en cuivre et d’autres maladies métaboliques rares mimant une MW (maladie de Menkes, acéruloplasminémie, syndrome CDG, maladie de Niemann-Pick de type C, syndrome MEDNIK). Par ailleurs, 20 % des hétérozygotes pour une mutation de l’ATPase 7B ont un taux de céruloplasminémie légèrement diminué entre 0,15-0,19 g/L. À l’inverse, la céruloplasminémie est augmentée en cas de syndrome inflammatoire ou d’état d’hyperœstrogénie, par exemple en cas de prise de contraception orale de type oestroprogestatif ou de grossesse.
La mesure de la cuprurie/24 heures est un des éléments clés du diagnostic de la MW. Il s’agit d’un test simple et sensible qui reflète le niveau de cuivre libre non lié à la céruloplasmine dans la circulation. Dans une étude réalisée chez 111 volontaires sains, le taux moyen de cuprurie/24 h était de 21 µg/24 h (0,34 µmol/24 h) (8). D’après les recommandations internationales, un taux supérieur à 100 µg/24 h (> 1,6 µmol/24 h) est très évocateur du diagnostic de MW chez un sujet non traité (9). Ce cut-off paraît toutefois trop élevé notamment chez les enfants présentant une atteinte hépatique ainsi qu’en cas de formes présymptomatiques et un cut-off de 40 µg/24 h (0,64 µmol/24 h) a été proposé avec une sensibilité de 79 % et une spécificité de 88 % (10). La mesure de l’augmentation de la cuprurie/24 h après prise de D-pénicillamine (test au Trolovol) peut être proposée dans certaines situations difficiles. Ce test a essentiellement été validé chez l’enfant et est de moins en moins utilisé depuis la mise en place du dosage du cuivre échangeable et de la mesure du REC. La mesure de la cuprurie/24 h est également utilisée pour la surveillance de l’efficacité des traitements de la MW.
L’anémie hémolytique à Coombs négatif est un des éléments classiques du diagnostic de la MW. Elle est observée chez 4 à 10 % des patients et est hautement évocatrice quand elle est associée à une hépatopathie inexpliquée ou en cas de mouvements anormaux. Elle peut être révélatrice de la maladie dans certains cas et le diagnostic de MW doit toujours être éliminé en cas d’anémie hémolytique à Coombs négatif survenant chez un enfant ou un adulte jeune. Chez les patients présentant une forme fulminante, une hémolyse marquée est souvent retrouvée (50-80 % des cas), liée au relargage massif de cuivre libre toxique par les hépatocytes lésés.
En cas d’hépatite fulminante, la triade classique hypocuprémie-hypocéruloplasminémie-hypercuprurie est souvent prise en défaut. Dans cette situation, il est fréquent d’observer des taux normaux voire augmentés de cuivre sérique lié au relargage de cuivre libre par les hépatocytes, le taux de céruloplasminémie peut être abaissé, normal voire augmenté et le dosage de cuprurie/24 h est habituellement très augmenté mais de manière non spécifique car il est fréquemment observé une hypercuprurie dans les hépatopathies sévères non wilsoniennes. Le dosage du cuivre échangeable est habituellement très élevé. Dans ce cas particulier, en plus de la recherche d’une anémie hémolytique à Coombs négatif, la mise en évidence d’une relative faible augmentation des transaminases (< 2 000 UI/L) avec un ratio AST/ALT > 1, des valeurs normales ou faiblement augmentées du taux de Phosphatases alcalines (PAL) contrastant avec une élévation importante de la bilirubine (avec notamment un ratio PAL/bilirubine totale (en mg/ dL) <4) est évocateur du diagnostic de MW (11).
L’atteinte histologique de la MW n’est pas spécifique. De façon précoce, on observe des lésions de stéatose micro et/ou macrovacuolaire. Ensuite, l’aspect est celui d’une hépatite chronique active avec un infiltrat de cellules mononuclées, ressemblant à celui qui est observé dans une hépatite chronique virale ou auto-immune. Enfin, des lésions de fibrose apparaissent évoluant progressivement vers une cirrhose mixte micro et macronodulaire avec stéatose périportale. Lors des poussées d’insuffisance hépatique, on note l’apparition de larges zones de nécrose hépatocytaire. La biopsie hépatique est surtout réalisée en cas de forme atypique ou dans les formes sévères afin d’éliminer les diagnostiques différentiels. Les colorations du cuivre (coloration à la rhodanine) permettent de voir tardivement l’accumulation de cuivre stocké dans les lysosomes. Si une biopsie hépatique est pratiquée pour suspicion de maladie de Wilson, il est important de réaliser un dosage du cuivre intrahépatique. Chez les patients non traités, un taux augmenté de cuivre intrahépatique supérieur à 250 µg/g (4 µmol/g) est évocateur du diagnostic de MW mais un taux plus faible voire normal n’exclut pas le diagnostic du fait de la distribution hétérogène des dépôts de cuivre intrahépatocytaires (5). Les hépatopathies cholestatiques et les cirrhoses peuvent être à l’origine de faux positifs.
Les anomalies ophtalmologiques les plus fréquemment observées en cas de maladie de Wilson sont l’anneau de Kayser-Fleischer et la cataracte en tournesol. L’anneau de Kayser-Fleischer consiste en un dépôt de cuivre en forme d’anneau dans la couche interne de la cornée au niveau de la membrane de Descemet mis en évidence à l’examen à lampe à fente. Il s’agit d’une atteinte bilatérale retrouvée chez plus de 90 % des patients présentant une atteinte neurologique et environ la moitié des patients avec atteinte hépatique (36 % des enfants) (5). Cette anomalie est liée à la présence d’un excès de cuivre libre dans la circulation. Elle est très évocatrice du diagnostic de MW mais n’est pas pathognomonique car peut être retrouvée en cas de défaut d’excrétion biliaire du cuivre notamment en cas de cholestase chronique. Cette anomalie peut régresser sous traitement de la MW. La cataracte en tournesol est un signe classique mais rare de la MW avec des dépôts de cuivre au niveau du cristallin. Cette atteinte n’affecte pas l’acuité visuelle et peut également régresser sous traitement.
L’IRM cérébrale est l’examen de neuro-imagerie le plus sensible pour le diagnostic de maladie de Wilson. Des hypersignaux symétriques des ganglions de la base, du thalamus et/ou du tronc cérébral en séquences T2 ou FLAIR sont très évocateurs d’une MW et retrouvés chez plus de 90 % des patients avec atteinte neurologique et environ 50 % des patients avec atteinte hépatique (5). Le signe classique de la “tête de panda” caractérisé par des hypersignaux T2 entourant les noyaux rouges et la substance noire est très spécifique de la MW mais seulement retrouvé chez une minorité de patients. Des signes d’atrophie cérébrale diffuse +/- associés à des hyposignaux en séquences T2/T2* et SWI de la substance grise en lien avec une accumulation du fer sont fréquemment retrouvés chez les patients atteints de MW. Dans une étude de validation d’un score pronostique de sévérité des lésions IRM réalisées chez 39 patients naïfs de traitement le sous-score de toxicité aiguë basé sur la distribution et l’intensité des anomalies inflammatoires s’améliorait avec le traitement (p = 0,02) mais n’était pas corrélé avec le score UWDRS à baseline et après 2 ans de traitement. Le sous-score de toxicité chronique basé sur le degré d’atrophie cérébrale et les hyposignaux T2/T2*/SWI était par contre corrélé avec le score UWDRS à baseline (r = 0,59, p = 0,005) et après 2 ans de traitement (r = 0,68, p < 0,001) (12).
Le diagnostic sera confirmé par la mise en évidence de mutations du gène ATP7B. La figure 1B présente les laboratoires de biologie moléculaire réalisant cette analyse génétique du gène ATP7B. L’analyse génétique n’est réalisée que si le patient (ou ses parents si le patient est mineur) a signé un consentement éclairé. Plus de 600 mutations ont été décrites. La recherche des mutations causales dans le gène ATP7B (locus 13q14.3.) est réalisée par un séquençage de type Sanger ou par un séquençage dit de nouvelle génération (NGS, Next Generation Sequencing). Cette dernière technique est plus exhaustive car elle permet de séquencer facilement la totalité des régions codantes, la région promotrice voire les régions non codantes du gène ATP7B. La recherche de mutations par séquençage de l’intégralité de la séquence codante et des jonctions intron-exon permet actuellement de confirmer le diagnostic de MW dans 98 % des cas (5). En l’absence de mariages consanguins, la majorité des patients est hétérozygote composite avec une mutation différente sur chaque allèle. Les recherches de corrélations génotypes-phénotypes sont restées jusqu’à présent décevantes. Du fait du nombre important de mutations existantes, l’analyse génétique peut durer plusieurs semaines mais ne doit pas faire retarder l’instauration d’un traitement des formes présentant les critères classiques de la MW.
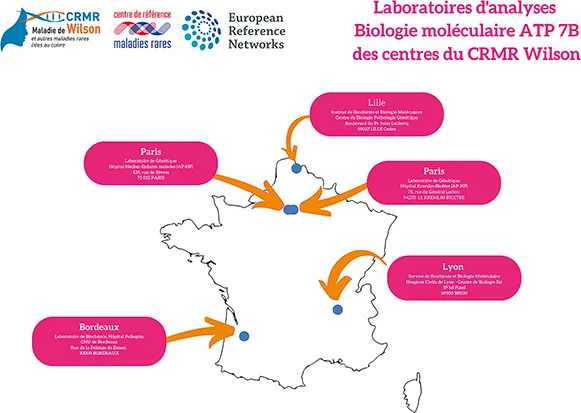
Figure 1B : Liste des laboratoires de biologie moléculaire des centres du CRMR Wilson
réalisant l’analyse du gène ATP7B (mise à jour décembre 2022)
En cas de diagnostic d’une MW, notamment en cas d’atteinte neurologique, des examens morphologiques hépatiques par échographie voire scanner et/ou IRM seront à réaliser systématiquement à la recherche de signes d’hépatopathie chronique (stéatose, hépatomégalie, dysmorphie hépatique, splénomégalie, ascite, réseau de dérivations collatérales, lésion focalisée).
Les données concernant les performances des tests non invasifs de fibrose dans la population de malades atteints d’une MW sont relativement rares. Dans une étude portant chez des sujets naïfs de traitement le seuil de 9,9 kPa a été proposé pour identifier les patients à risque de cirrhose (5). Les valeurs d’élasticité peuvent s’améliorer sous traitement comme observé dans d’autres hépatopathies.
Enfin, il est probable que l’on puisse s’appuyer sur les recommandations de BAVENO VII pour le dépistage d’une hypertension portale cliniquement significative bien qu’aucune étude n’a été réalisée spécifiquement dans la population de malades atteints d’une MW.
Il est essentiel de réaliser un dépistage familial en cas de nouveau diagnostic de MW. Le dépistage des apparentés inclut un examen clinique, un bilan biologique avec dosage de la céruloplasmine sérique, de la cuprurie des 24 heures, de la cuprémie totale et du cuivre échangeable (pour calcul du REC), des enzymes hépatiques et une recherche en biologie moléculaire des mutations familiales du gène ATP7B par un séquençage Sanger des exons concernés (5).
Le dépistage est à réaliser en priorité dans la fratrie, chaque frère et sœur du cas index ayant un risque de 25 % d’être atteint (homozygote ou hétérozygote composite). Les parents du cas index sont aussi dépistés afin de confirmer que les mutations trouvées sont bien en trans et d’éliminer des cas de pseudo-dominance.
Étant donné la prévalence génétique et l’existence d’un traitement efficace, il est également recommandé de proposer un dépistage chez les oncles, tantes et cousins/ cousines du cas index.
Enfin, pour les enfants du cas index, le dépistage sera proposé après l’âge de trois ans pour le diagnostic des formes présymptomatiques.
Il doit être entrepris le plus rapidement possible et maintenu à vie. Les traitements médicamenteux spécifiques de la MW sont les chélateurs oraux du cuivre (D-pénicillamine, Trientine) et le zinc (tableau 3).
| Chélateurs | Sels de Zinc | ||
|---|---|---|---|
| D-Pénicillamine | Trientine | Acétate de Zinc | |
| Trolovol® cp 300mg | Cufence® (TETA-2HCL) gél 200mg Cuprior® (TETA_4HCL) cp 150 mg | Wilzin® gél 50mg | |
| Mode d’action | |||
| Élimination urinaire du cuivre | ↑↑↑ | ↑↑ | – |
| Absorption digestive du cuivre | – | ↓ | ↓↓ |
| Posologie (chez l’adulte) | |||
| Attaque | 1200 à 1800 mg/j | Cufence 800 à 1600 mg Cuprior 600 à 1050 mg | 100 à 150 mg/j |
| Entretien | 600 à 1200 mg | Cufence 400 à 800 mg Cuprior 450 à 900 mg | 100 à 150 mg/j |
| Indication | |||
| 1ère ligne des formes symptomatiques (hépatique ou neurologique) | 2ème ligne si effets secondaires ou échappement 1ère ligne si allergie pénicilline | 1ère ligne des formes présymptomatiques 2ème ligne si effets secondaires ou échappement | |
| Surveillance | |||
| Cuivre échangeable | ↓ | ↓ | ↓ |
| Cuprurie/24h | ↑↑ | ↑ | ↓ |
| Tolérance | |||
| Risque d’aggravation neurologique | ++(+) | ++ | + |
| Toxicité | +++ | + | + |
| Effets secondaires | Cutané, hématologique, rénal, rhumatologique, maladies auto- immunes | Hématologique, digestif | Digestif |
En cas de forme symptomatique, le traitement chélateur initial visera à obtenir dans un premier temps une déplétion de la surcharge cuprique de l’organisme (traitement d’attaque d’une durée en général de 6 à 24 mois) puis dans un second temps à prévenir la reconstitution d’une surcharge cuprique (traitement d’entretien à vie).
Lors de la phase initiale chélatrice, il est habituel de recommander au patient de suivre un régime pauvre en cuivre en évitant les aliments riches en cuivre tels que le chocolat noir, le foie, les crustacés, les champignons, les fruits secs, ou encore les noix et noisettes (tableau 4). Une attention particulière devra être portée aux compléments alimentaires oraux thérapeutiques parfois très riches en cuivre. Ce régime pourra être moins stringent lors de la phase d’entretien.
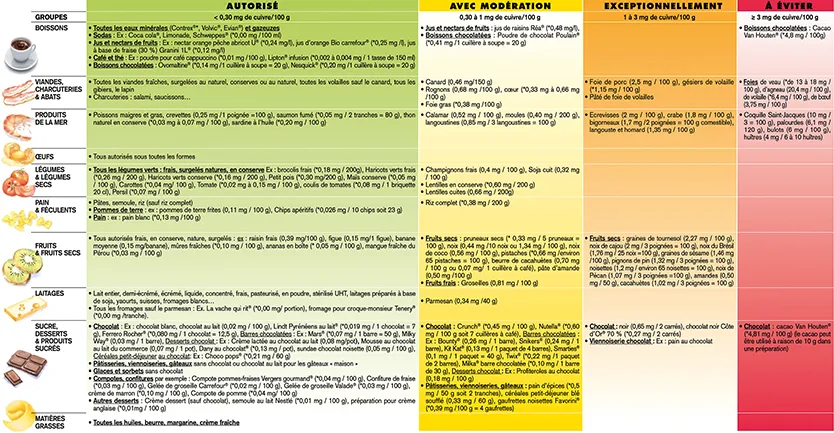
Tableau 4 : Liste de différents aliments selon leur teneur en cuivre
La D-pénicillamine est le traitement de référence (5). Il s’agit du chélateur le plus puissant mais également potentiellement le plus toxique. Ce traitement permet l’élimination du cuivre par voie urinaire. Il est indiqué en première intention pour le traitement des formes symptomatiques (hépatiques ou neurologiques). Il existe un risque d’aggravation clinique paradoxale lors des premières semaines de traitement notamment de l’atteinte neurologique. Pour cette raison, la dose doit être progressivement croissante, en débutant à 150 mg (chez enfant) et 300 mg chez l’adulte puis en augmentant toutes les semaines (voire tous les mois en cas d’atteinte neurologique). Son efficacité est jugée par la mesure de la cuprurie des 24 heures qui peut atteindre initialement des chiffres très élevés (habituellement >1 000 µg/j (16 µmol/j)) les premiers mois de traitement puis avec un objectif de cuprurie entre 200 et 500 µg/j (3 à 8 µmol/j) durant la phase de traitement d’entretien, et par le dosage du cuivre échangeable qui doit se normaliser (9,13,14). La cytolyse doit aussi régresser progressivement en six à douze mois après le début du traitement. La posologie en traitement d’attaque est habituellement chez l’adulte de 900 à 1 800 mg/j en 2 à 3 prises à prendre à distance des repas (30 minutes à 1 heure avant et 2 heures après) puis 600 à 1 500 mg/j pendant la phase d’entretien. Chez l’enfant, la posologie usuelle initiale est de 20 à 30 mg/kg/j (1 800 mg/j au maximum) en 2 à 4 prises puis de 10 à 20 mg/kg en traitement d’entretien. Durant le suivi, environ 30 % des patients doivent interrompre la D-pénicillamine en raison d’effets secondaires : réactions d’hypersensibilité, myélotoxicité, néphrotoxicité avec protéinurie isolée voire syndrome néphrotique, développement de maladies auto-immunes dont le lupus induit, lésions cutanéo-muqueuses (aphtose buccale, élastopathie).
La trientine a aussi des propriétés chélatrices du cuivre libre en augmentant son excrétion urinaire couplées à une diminution de l’absorption intestinale du cuivre. Il existe actuellement 2 formes de Trientine : TETA-2HCL (Cufence®), TETA-4HCL (Cuprior®) (5).
La dose recommandée de Cufence® 200 mg chez l’adulte varie de 800 à 1 600 mg/j (4 à 8 gélules) par jour fractionnée en 2 à 4 prises, et chez l’enfant, la dose est de 20 mg/kg/j (max de 1 500 mg/j) en 2 à 4 prises.
Concernant le Cuprior® 150 mg, la dose proposée chez l’adulte est de 450 mg à 1 050 mg par jour (3 à 7 comprimés pelliculés) fractionnée en 2 à 4 prises. Chez l’enfant de plus de cinq ans, la dose est généralement de 300 mg à 600 mg par jour (2 à 4 comprimés pelliculés) fractionnée en 2 à 4 prises.
La survenue d’effets indésirables liés à la Trientine est plus rare qu’avec la D-Pénicillamine : anémie sidéroblastique réversible en cas de forte dose (liée à la chélation du fer), cytopénies voire rare lupus induit. Des troubles digestifs (diarrhée/ constipation, douleurs abdominales) et cutanées (sécheresse cutanée, prurit intermittent) ont été décrits.
La trientine est un traitement de deuxième intention utilisé en cas de survenue d’effets indésirables à la D-pénicillamine ou aux sels de Zinc ou en cas d’échappement thérapeutique à l’une de ces molécules. Ce traitement peut également être utilisé en première intention en cas de contre-indication à la D-pénicillamine.
L’efficacité du traitement par sel de TETA est jugée sur l’élévation de la cuprurie des 24 heures (plus modeste que sous D-Pénicillamine) et par le dosage du cuivre échangeable qui doit se normaliser. Les transaminases doivent également se normaliser.
Les sels de zinc ont un mode d’action différents des chélateurs oraux : ils bloquent l’absorption intestinale du cuivre en induisant la synthèse d’une métallothionéine intestinale, qui a plus d’affinité pour le cuivre que pour le zinc et accroît son excrétion fécale. Dans le foie, ils induisent également la synthèse de métallothionéines fixant le cuivre et empêchant son passage dans le sang (5).
Le sel de Zinc le plus utilisé est le Wilzin®. La posologie habituelle chez l’adulte est de 150 mg/j en 3 prises à distance des repas. Chez l’enfant, la posologie dépend de l’âge et du poids et varie 25 mgx2/j (avant 6 ans) à 50 mgx3/j après 16 ans pour un poids de plus de 57 kg. En cas d’intolérance gastrique, survenant fréquemment lors la prise du matin, cette prise peut être retardée jusqu’au milieu de la matinée, entre le petit déjeuner et le déjeuner. Il est également possible de prendre Wilzin® avec un peu de protéines.
L’objectif à terme de ce traitement est d’obtenir une normalisation du cuivre échangeable et une diminution de la cuprurie/24 heures avec des seuils < 75-100 µg/j (< 1,2 à 1,6 µmol/j). Il s’agit du traitement de première intention des formes présymptomatiques. Il peut également être utilisé en 2e intention en cas d’intolérance ou d’échappement thérapeutique à la D-pénicillamine et à la Trientine.
La surveillance de l’efficacité des traitements est basée sur l’amélioration des signes cliniques de décompensation hépatique, des symptômes neurologiques (avec suivi de l’évolution du score UWDRS), des tests hépatiques et des fonctions de synthèse hépatique. En parallèle, la surveillance de la cuprurie/24 heures couplé au dosage du cuivre échangeable sera réalisé régulièrement. Les objectifs de cuprurie dépendent de la phase de traitement (initial vs. entretien) et du type de traitement (chélateurs vs. sels de zinc). Le suivi du cuivre échangeable représente également un outil intéressant pour le suivi de l’efficacité des traitements mais les seuils optimaux d’efficacité restent à déterminer.
La transplantation hépatique permet de corriger l’anomalie métabolique en rétablissant une excrétion biliaire par le greffon qui est porteur d’une ATPase 7B fonctionnelle. Les indications classiques de transplantation hépatique chez les patients présentant une MW sont l’hépatite fulminante révélant la MV ou compliquant un arrêt prolongé de traitement et la cirrhose décompensée ne répondant pas au traitement médical spécifique (le plus souvent chélateur) (figure 2) (15,16). En cas d’hépatite aigue sévère, la détermination du score NWI (New Wilson Index ou score de Nazer modifié) permet d’aider à la décision thérapeutique (tableau 5). Le NWI correspond à un score pronostique regroupant cinq variables (bilirubine totale, INR, ASAT, globules blancs, albuminémie) dont deux communes avec le MELD, développé initialement dans une cohorte pédiatrique (17). Un score supérieur à 10 est associé à un risque de mortalité accru mais n’est pas une contre-indication à l’instauration d’un traitement médical spécifique (en l’absence d’encéphalopathie hépatique) sous surveillance rapprochée stricte dans un centre de transplantation hépatique. Les indications de greffe pour carcinome hépatocellulaire, complication relativement rare chez les patients atteints de MW, suivent le score AFP. Le pronostic post-greffe immédiat et à long terme des patients atteints de MW est bon. Dans la cohorte française de 121 patients transplantés entre 1985 et 2009, les taux de survie des patients étaient à 89 % à 1 an et 87 % à 5, 10 et 20 ans. Les taux de survie des greffons étaient à 81 % à 1 an, 80 % à 5 ans, 79 % à 10 ans et 70 % à 20 ans (18). Le jeune âge des patients, l’absence de comorbidités, l’absence de récidive de la maladie initiale sur le greffon et la faible proportion de patients greffés pour un carcinome hépatocellulaire expliquent ces bons résultats. L’indication de greffe pour atteinte neurologique sévère ne répondant pas au traitement ou s’aggravant sous traitement ne fait pas l’objet d’un consensus international et reste débattue, bien que des données récentes rapportent des résultats intéressants (19). En France, cette indication est discutée au cas par cas entre le centre de transplantation et le centre de référence pour la maladie de Wilson.
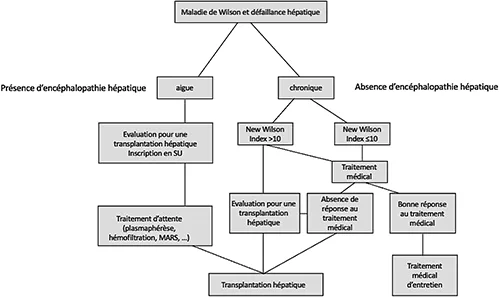
Figure 2 : Algorithme décisionnel en cas de maladie de Wilson avec défaillance hépatique (adapté d’après Ahmad et al., Handbook of Clinical Neurology, 2017 (Chapitre 16 Liver transplantation for Wilson disease)
| Score | Bilirubine (µmol/L) | INR | ASAT (UI/L) | GB (109/L) | Albumine (g/L) |
|---|---|---|---|---|---|
| 0 | 0-100 | 0-1,29 | 0-100 | 0-6.7 | >44 |
| 1 | 101-150 | 1,3-1,6 | 101-150 | 6,8-8,3 | 34-44 |
| 2 | 151-200 | 1,7-1,9 | 151-300 | 8,4-10,3 | 25-33 |
| 3 | 201-300 | 2,0-2,4 | 301-400 | 10,4-15,3 | 21-24 |
| 4 | >300 | >2,4 | >400 | >15,3 | <21 |
Concernant les traitements non spécifiques de l’atteinte hépatique, les modalités de prise en charge de l’hypertension portale cliniquement significative s’appuient sur les recommandations de BAVENO VII. Une vaccination prophylactique contre le virus de l’hépatite A et l’hépatite B seront à proposer au patient ainsi qu’une vaccination prophylactique contre la grippe, la COVID et le pneumocoque en cas de cirrhose.
Concernant les traitements non spécifiques de l’atteinte neurologique, des traitement sont proposés au cas par cas pour la prise en charge des symptômes neuropsychiatriques : dystonie (anticholinergique, toxine botulique), tremblement (B-bloquant), mouvements choréiques (neuroleptique atypique), spasticité (baclofène), hypersalivation (anticholinergique, toxine botulique), syndrome dépressif (IRS, tricycliques), troubles psychotiques (neuroleptique atypique), épilepsie symptomatique (antiépileptiques). En parallèle une prise en charge paramédicale (kinésithérapie, ergothérapie, orthophoniste, psychologique, neuropsychologique) sera associée (5).
L’adhérence est la clé du succès thérapeutique et représente un véritable défi chez les patients atteints d’une MW, souvent jeunes, parfois asymptomatiques, afin d’éviter la survenue d’une aggravation de l’atteinte existante ou l’apparition de nouveaux symptômes. Des études récentes rapportent des problèmes d’adhérence chez près d’un tiers des patients particulièrement en cas de formes hépatiques ou présymptomatiques (20).
MW : Maladie de Wilson
UWDRS : Unified Wilson Disease Rating Scale
REC : Ratio cuivre échangeable/cuivre total
NGS : Next Generation Sequencing
NWI : New Wilson Index
FMC HGE : Organisme certifié Qualiopi pour la catégorie ACTIONS DE FORMATION.
