Liens d’intérêt
Gilead (Orateur), AbbVie (Orateur), Novartis (Expertise), La Jolla (Expertise)
Mots-clés
Hémochromatose, HFE, fer
Abréviations
Non communiquées
Introduction
L’hémochromatose est la maladie génétique la plus fréquente dans la population française touchant potentiellement une personne sur 250. Décrite et bien connue depuis plusieurs décennies, l’amélioration des connaissances scientifiques a permis une nette progression de sa prise en charge, mais a aussi soulevé la complexité de son expression. Le champ diagnostic génétique s’est considérablement élargi, tandis qu’à l’inverse la variabilité des conséquences cliniques chez des patients « porteurs » sur le plan génétique a été mise en évidence. Deux efforts majeurs de consensus effectués récemment ont cherché d’une part, à clarifier la définition et la classification de l’hémochromatose, porté par la société savante BioIron (International Society for the Study of Iron in Biology and Medicine), et d’autre part à répondre de façon pragmatique et consensuelle aux questions que pose la prise en charge des patients, portée par la société savante EASL (European Association for the Study of the Liver).
Définition et classification de l’hémochromatose
La BIOIRON society est la société savante internationale s’intéressant au métabolisme du fer. Elle regroupe les scientifiques de différents horizons (biologie moléculaire, biochimie, physique), les cliniciens (hépatologues, hématologues, biologistes, radiologues, néphrologues, neurologues, internistes) travaillant de façon transversale sur tous les axes du métabolisme du fer. Au fil des avancées scientifiques mettant en évidence les différents acteurs impliqués dans la régulation du métabolisme du fer, la définition de l’hémochromatose et de ses différentes formes a évoluée. Il est apparu un décalage entre la complexité fondamentale et la prise en charge pragmatique des patients. Un travail de consensus fut initié en 2019 dans l’objectif de définir ce qu’était une hémochromatose, et quelle pouvait être la classification simple sur le plan clinique, mais cohérente sur le plan scientifique pour en distinguer les différentes formes. Le fruit de ce travail a été publié en 2022 (1).
Définition de l’hémochromatose
L’hémochromatose est une maladie du métabolisme du fer caractérisée par une absorption digestive dérégulée. La conséquence est l’augmentation du fer circulant avec un coefficient de saturation de la transferrine élevé puis une possible surcharge organique en fer, en particulier hépatique. Cette approche clinique a par la suite été complétée par les données de la génétique, avec la découverte du gène HFE. 80 % des cas d’hémochromatose dans la population européenne sont liés à une homozygotie p.C282Y (2, 3). La découverte de nombreux facteurs régulateurs et transporteurs ont permis la meilleure compréhension du métabolisme du fer. L’hepcidine et les facteurs associés à sa régulation, dont HFE fait partie, se sont imposés comme les pivots de la pathologie avec une physiopathologie et une présentation clinique cohérente.
Il est donc maintenant considéré qu’une hémochromatose correspond à une anomalie génétique conduisant à un déficit en hepcidine altérant l’équilibre de l’axe hepcidine-ferroportine. Ne sont ainsi considérées comme hémochromatose que les formes de surcharge en fer d’origine génétique, excluant toute cause acquise (1) (Tableau 1). L’absence de cause acquise doit être considérée de façon proportionnelle par rapport à la sévérité de la surcharge et du contexte clinique. La présence associée de plusieurs éléments du syndrome métabolique (hypertension, surpoids, diabète, dyslipidémie) peut ainsi facilement expliquer une hyperferritinémie ou une surcharge en fer modérée chez un homme âgé (hépatosidérose métabolique). À l’inverse chez une femme jeune, la simple présence isolée d’un des éléments du syndrome métabolique explique plus difficilement une concentration hépatique en fer ou une ferritine à plusieurs fois la normale. Les causes acquises sont donc à discuter au cas par cas pour évaluer les seuils incitant à pousser les explorations génétiques.
L’hémochromatose est caractérisée sur le plan clinique par une augmentation du coefficient de saturation de la transferrine (>45 % à deux reprises), une surcharge en fer hépatique (>100-120 µmol/g), l’absence de surcharge splénique, associée ou non à des signes cliniques compatibles.
Les pathologies hématologiques génétiques, (par exemple les thalassémies ou myélodysplasies) peuvent être associées à une surcharge en fer même en l’absence de transfusion. Pour éviter la confusion entre l’hémochromatose et ces pathologies génétiques hématologiques, l’absence d’anomalie de la lignée rouge (anémie ou réticulocytose) est indispensable au diagnostic.
Classification de l’hémochromatose
La mise en évidence des nouveaux acteurs du métabolisme du fer a conduit à une classification de l’hémochromatose en fonction de la caractérisation génétique. Ces progrès ont permis de réduire la part de surcharge non expliquée, mais ont également abouti à une classification peu pratique sur le plan clinique (4). Les différentes formes sont classées par un chiffre dépendant de l’ordre chronologique de découverte du gène, puis pour certain en sous type A/B en fonction de variations phénotypiques.
Cette classification intéressante sur le plan moléculaire présente de nombreuses limites. En premier lieu les tests génétiques de deuxième ligne ne sont pas disponibles dans tous les centres, et demandent une interprétation complexe pour certains variants. Tous les patients ne peuvent donc être classés et la classification ne doit pas retarder la prise en charge thérapeutique. Par ailleurs les phénotypes initialement décrits comme homogènes se sont révélés beaucoup moins cohérents avec l’extension du dépistage de ces formes, avec par exemple des formes « juvéniles » mises en évidence chez des patients de plus de 60 ans. Le nombre toujours plus important de gènes impliqués nécessiterait une extension numérique continue de la classification. Enfin certaines données suggèrent que des formes de surcharge puissent être liées à l’association de variants délétères à l’état hétérozygote dans différents gènes (digénisme, cf. infra).
Le groupe de travail a donc proposé une nouvelle classification se voulant plus pertinente sur le plan clinique et plus ouverte à l’évolution des connaissances (Tableau 2).
Les points principaux de cette classification outre la simplification, sont en premier lieu l’uniformisation du phénotype autour du déficit en hepcidine, et la mise en avant d’une hémochromatose HFE élargie puisque prenant désormais en compte les variants rares du gène HFE. Sur ce point il est aussi fortement souligné l’importance de caractériser les variants de façon adaptée pour ne pas retenir de variant comme délétère à tort. De la même façon, il est nécessaire de discuter en RCP le diagnostic de digénisme, devant la présence de plusieurs variants délétères à l’état hétérozygote, mais présents en association sur des gènes distincts impliqués dans la même cascade de régulation. Les hémochromatoses non liées à HFE et digéniques laissent la porte ouverte aux futurs acteurs identifiés du métabolisme du fer. Enfin l’ancien sous type A de la Ferroportine est exclu du champ des hémochromatoses, son phénotype et son mécanisme particulier en faisant une entité à part entière, la maladie de la Ferroportine.
Tableau 1 : critère diagnostiques d’une hémochromatose
| Critères diagnostiques d’une hémochromatose |
| Saturation de la transferrine > 45 % | (contrôlée à deux reprises) |
| Surcharge hépatique en fer | (évaluée par IRM) |
| Absence de surcharge macrophagique/splénique |
| Absence de maladie hématologique | |
| Absence de cause acquise de surcharge en fer | Alcool Syndrome métabolique Maladie chronique du foie Transfusions (significative par rapport au niveau de surcharge) |
Tableau 2 : Classification des hémochromatoses
| Nouvelle Classification | Génétique | Commentaires |
| Liée à HFE | p.C282Y homozygote heterozygote composite : p.C282Y avec variant rare délétère du gène HFE | Le variant p.H63D n’est pas un variant délétère et ne peut expliquer un diagnostic. L’interprétation des variants non connus doit être prudente et faire l’objet de discussion en RCP |
| Non liée à HFE | Variant délétère dans un gène autre que HFE – HJV – HAMP – TFR2 – SLC40A1 Gain de Fonction | Potentiellement tous les gènes impliqués dans la régulation du métabolisme de l’hepcidine peuvent être responsables. Les nouveaux gènes candidats doivent faire l’objet de validations in vitro et épidémiologiques |
| Digénique | Double hétérozygotie ou homozygotie pour des variants délétères dans 2 gènes différents du métabolisme du fer | Principalement l’association du variant p.C282Y du gène HFE, à un ou des variants délétères dans un autre gène du métabolisme du fer |
| Génétiquement indéterminée | Caractérisation génétique non (encore) disponible après séquençage des gènes connus | Dans la mesure du possible, adresser le patient à un centre expert pour poursuite des explorations |
HJV : Hémojuvéline, HAMP : hépcidine, SLC40A1 : ferroportine, TFR2 : récepteur 2 de la transferrine
Recommandations européennes pour la prise en charge des hémochromatoses
Dans le cadre de la mise à jour de ses recommandations de pratique clinique, l’EASL a souhaité modifier leur structure pour s’orienter vers une méthodologie de type PICO/Delphi. Les précédentes recommandations de 2010, de méthodologie plus classique, visaient à proposer un consensus : étude bibliographique extensive afin de mettre en avant les preuves disponibles, un consensus d’expert était proposé sur ces faits, puis un niveau de preuve était attribué selon la classification GRADE. Dans ces nouvelles recommandations, un panel d’experts a défini 15 questions cliniques pour la prise en charge des patients, qui ont été validées par un panel d’experts externe dans le cadre d’une méthode Delphi. Puis le panel d’experts a rédigé un consensus basé sur la littérature et pragmatique pour répondre à ces questions. Ensuite les réponses ont fait l’objet d’une validation par le panel externe. Ces recommandations se veulent donc plus pragmatiques et orientées vers la pratique clinique.
Dépistage et diagnostic
Qui devrait être dépisté pour l’hémochromatose ?
Les signes cliniques associés à l’hémochromatose sont dépendants du stade de la maladie et du niveau de surcharge en fer. L’asthénie et les douleurs articulaires qui sont les signes cliniques les plus fréquents sont peu spécifiques. Dans les formes les plus sévères ou les plus avancées, les signes cliniques de maladie chronique du foie, une arythmie cardiaque, une mélanodermie et/ou un trouble endocrinien sont possibles (5).
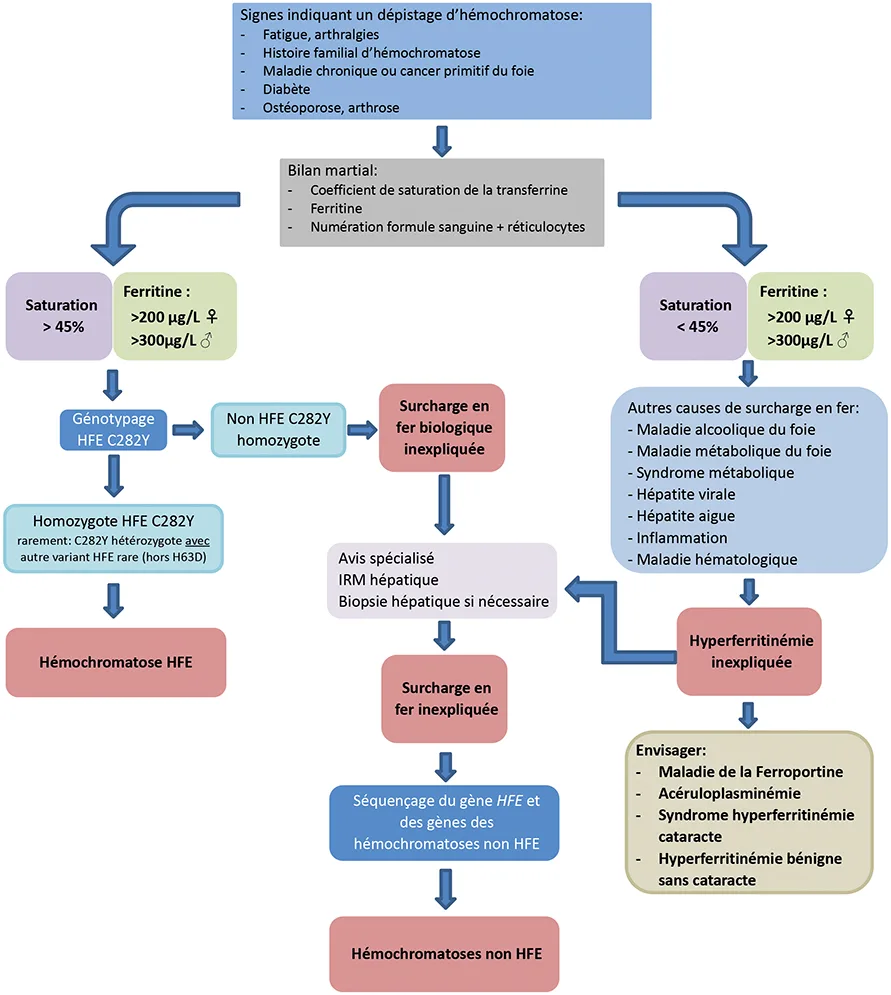
Biologiquement les signes devant faire évoquer une hémochromatose sont l’association d’un coefficient de saturation de la transferrine élevé (>45 % chez la femme, 50 % chez l’homme) ainsi qu’une augmentation de la ferritine (>200 µg/L chez la femme et >300 µg/L chez l’homme) (Figure 1). Une surcharge en fer peut par ailleurs être mise en évidence de manière fortuite sur une biopsie ou une IRM hépatique.
Tous les patients présentant des signes cliniques ou biologiques évocateurs d’hémochromatose, ou majeurs apparentés au 1er degré d’un patient avec une hémochromatose doivent être dépistés pour l’hémochromatose.
Quel doit être le bilan biologique de première ligne pour dépister l’hémochromatose ?
Le premier signe biologique de l’hémochromatose est l’augmentation du coefficient de saturation de la transferrine. En raison de la variabilité de son dosage, un contrôle sur un deuxième prélèvement est souhaitable. Elle peut être faussement augmentée en cas de dysérythropoïèse ou d’insuffisance hépatocellulaire. Le deuxième signe biologique est l’augmentation de la ferritine. Néanmoins la ferritine peut être augmentée dans de nombreuses situations (inflammation, syndrome métabolique, alcool, etc.) qui doivent donc être considérées pour son interprétation (Figure 1). Bien que central sur le plan physiologique, le dosage de l’hepcidine n’a aucun intérêt clinique.
Le bilan biologique d’une hémochromatose doit comporter la mesure du coefficient de saturation de la transferrine et de la ferritine.
Qui devrait être testé pour le variant p.C282Y du gène HFE ?
L’homozygotie p.C282Y explique la vaste majorité des cas d’hémochromatoses dans la population caucasienne (>80 %). La prévalence de ce variant présente un gradient Nord-Sud en Europe, avec une prévalence estimée en France à 6,4 % de la population (soit une personne homozygote p.C282Y sur 250) (Figure 1). Néanmoins la pénétrance de l’homozygotie p.C282Y est variable en fonction du sexe et de l’âge, ainsi le dépistage systématique en population n’est pas considéré comme pertinent.
Un génotypage p.C282Y HFE devrait être proposé aux patients caucasiens présentant des signes biologiques de surcharges en fer, avec ou sans signes cliniques, ainsi qu’aux majeurs apparentés au 1er degré de patients avec une hémochromatose HFE.

Figure 1
Les patients devraient-ils être aussi testés pour le variant p.H63D ?
La prévalence du variant p.H63D dans la population est très élevée, ce qui est en faveur d’un variant bénin. De grandes études de population ont montré que le variant p.H63D à l’état homozygote, ou hétérozygote composite avec le variant p.C282Y n’avait aucune conséquence clinique (6). Les cas décrits de surcharge en fer, associé à ce génotype, ont de façon quasi systématique des surcharges modérées, et présentent plusieurs cofacteurs pouvant expliquer la surcharge. La présence de ce variant ne peut expliquer une surcharge significative, et les explorations génétiques complémentaires doivent donc être poursuivies. En France la réalisation de ce test n’est pas recommandée et n’est pas prise en charge par la Sécurité sociale.
La réalisation d’un génotypage p.H63D n’est pas recommandé. Les patients homozygotes p.H63D ou hétérozygotes composite p.C282Y/H63D présentant une surcharge en fer doivent être explorés pour les autres causes génétiques ou acquises de surcharge en fer.
Quand devrait être réalisée une recherche des variants génétiques rares d’hémochromatose ?
Si la recherche de l’homozygotie p.C282Y est le premier test à effectuer pour le diagnostic d’une hémochromatose en population caucasienne, cette attitude est moins pertinente dans les populations non caucasiennes ou sa prévalence est faible (<1-2/10 000). Il a été clairement établi que l’absence de diagnostic précis engendrait de nombreuses conséquences psychologiques ayant un impact direct sur la qualité de vie et la prise en charge du patient. (7).
Chez les patients présentant une hémochromatose sans homozygotie p.C282Y la recherche de variant rare est donc recommandée pour limiter les conséquences de l’incertitude, améliorer l’évaluation pronostique et guider les explorations complémentaires, permettre un dépistage familial, et améliorer la compréhension des anomalies du métabolisme du fer.
En France, pour discuter de la réalisation et interprétation de telle recherche, il est recommandé de se rapprocher d’un centre du réseau maladie rare des hémochromatoses (www.centre-reference-fer-rennes.org). Il n’existe pas de donnée permettant d’établir des valeurs seuil claires à partir desquelles ces recherches génétiques doivent être engagées. Il est en effet indispensable de prendre en compte le phénotype, les cofacteurs personnels et environnementaux éventuels, et l’histoire familiale. L’interprétation des résultats concernant les variants identifiés peut être délicate et doit faire l’objet de discussion en RCP (Figure 1).
La recherche des variants rare doit comporter au minimum l’analyse des gènes HFE, HAMP, HJV, TFR2, TF, CP, BMP6 et SLC40A1. En cas de négativité la réalisation d’un exome, permettant d’élargir les recherches au-delà des gènes classiquement associés au métabolisme du fer, doit être discutée avec un centre expert si elle est disponible.
Les patients jeunes présentant une hémochromatose avec des manifestations cliniques organiques doivent être testés pour les variants rares des gènes de l’hémochromatose. Les dossiers de patients présentant une hémochromatose inexpliquée doivent être discutés avec un centre expert pour la recherche des variants rares des gènes de l’hémochromatose. Les apparentés au 1er degré et majeurs de patients présentant des variants rares considérés comme délétères des gènes de l’hémochromatose doivent être dépistés pour ces variants.
Explorations complémentaires
Chez quels patients une quantification non invasive de la charge en fer devrait être proposée ?
Les marqueurs biologiques du métabolisme du fer sont des reflets indirects de la charge en fer de l’organisme. La saturation de la transferrine peut être augmentée par une consommation d’alcool excessive ou une insuffisance hépatocellulaire. La ferritine peut être augmentée par un syndrome inflammatoire indépendamment de sa cause, une lyse cellulaire, le syndrome métabolique ou une consommation d’alcool. Les marqueurs biologiques sont donc parfois insuffisants pour évaluer le stock en fer de l’organisme. La concentration hépatique en fer peut être estimée de façon non invasive par IRM, le coût et la disponibilité de cet examen peuvent néanmoins limiter son recours. Chez un patient présentant une hémochromatose HFE sans cofacteur, la mesure de la concentration hépatique en fer est inutile car la ferritine est alors un bon reflet du stock en fer. Chez un patient sans diagnostic précis, ou présentant des cofacteurs rendant l’interprétation des marqueurs biologiques délicats, la mesure de la concentration hépatique en fer aide à guider la prise en charge.
Chez les patients avec une cause incertaine d’hyperferritinémie ou de surcharge en fer, une IRM hépatique devrait être proposée pour quantifier la concentration hépatique en fer et évaluer le risque de surcharge en fer extrahépatique. Une IRM cardiaque peut être proposée aux patients présentant une hémochromatose et une cardiopathie, ainsi que dans les formes juvéniles d’hémochromatoses.
Quand est-il nécessaire de proposer une biopsie hépatique chez un patient avec une hémochromatose ?
Avec le développement des outils non invasifs pour mesurer le stock en fer, la biopsie hépatique n’a d’utilité que pour évaluer la sévérité de la fibrose hépatique, ou faire la part des choses lorsque plusieurs maladies du foie sont intriquées. La sévérité de la fibrose étant proportionnelle au niveau de surcharge en fer, le risque de fibrose est négligeable si la ferritine est inférieure à 1 000 µg/L. Ce seuil prudent peut toutefois être modulé par la présence d’autres maladies du foie associée (syndrome métabolique, alcool). La biopsie hépatique peut ainsi être discutée en cas de ferritine inférieure à 1 000 µg/L si elle est associée à une cytolyse. Les différents tests non invasifs biologiques ou physique (élastométrie), bien qu’intéressants dans le contexte, n’ont pas encore été validés à ce jour. Chez un patient présentant une hémochromatose, si la cirrhose ne peut être écartée ou affirmée avec certitude, une biopsie hépatique doit être proposée.
La biopsie hépatique vise à évaluer la présence d’une cirrhose, elle n’est donc pas recommandée en cas de diagnostic de cirrhose évident. La biopsie hépatique n’est pas recommandée pour évaluer la charge hépatique en fer. Une biopsie peut être proposée pour évaluer la fibrose si la ferritine est supérieure à 1 000 µg/L ou s’il existe une cytolyse.
Quand et comment le degré de fibrose doit être évalué ?
Des travaux préliminaires ont montré des résultats intéressants concernant les tests non invasifs de fibrose (APRI, FIB-4, élastométrie hépatique), cependant il s’agissait d’études monocentriques et d’effectifs modestes limitant la force de leurs conclusions. Il semble néanmoins que du fait de sa physiopathologie particulière, les seuils soient significativement plus bas que pour les autres maladies chroniques du foie (hépatite virales, maladie du foie lié à l’alcool). Aucune donnée scientifique ne permet d’établir à quel rythme le degré de fibrose doit être évalué au-delà du diagnostic initial.
Tous les patients ayant une hémochromatose doivent avoir une évaluation de la fibrose lors du diagnostic initial. L’élastométrie hépatique peut permettre d’éliminer une fibrose significative si elle est <6.4kPa. Les données sont limitées concernant les scores biologiques (FIB-4).
Quelles manifestations extra hépatiques doivent être recherchées ?
Les arthropathies et l’ostéoporose sont fréquentes chez les patients avec une hémochromatose, et ne répondent pas de façon uniforme au traitement par saignée. La prévalence des douleurs articulaires et des remplacements prothétiques est particulièrement élevée, affectant principalement les mains, poignets, chevilles et hanches (6). Les manifestations articulaires sont similaires à celle de l’arthrose classique, mais présentes à un âge plus jeune, avec des ostéophytes et une perte du cartilage plus rapide. L’arthropathie ne répond pas toujours au traitement, et peut même apparaître ou progresser chez les patients sous traitement, elle présente un impact très significatif sur la qualité de vie. Son traitement se limite à la prise en charge symptomatique rhumatologique.
Dans les hémochromatoses sévères, une atteinte endocrinienne ou cardiaque est présente. C’est particulièrement le cas dans les formes juvéniles, pouvant engendrer des insuffisances cardiaques majeures (5). La surcharge en fer myocardique peut entraîner une cardiopathie restrictive avec dysfonction diastolique, pouvant évoluer vers une cardiopathie dilatée avec dysfonction systolique associée. La surcharge en fer peut aussi affecter le tissu nodal, en particulier le nœud atrio-ventriculaire, occasionnant différents types de trouble du rythme. L’atteinte cardiaque doit être évaluée de façon classique, au-delà de l’examen clinique, par une échographie cardiaque transthoracique ainsi qu’un électrocardiogramme. Une IRM cardiaque avec quantification du fer cardiaque peut être intéressante lorsqu’elle est disponible. Il n’existe pas de seuil défini de ferritine ou de concentration hépatique en fer qui soit associé avec l’apparition d’une surcharge cardiaque. Pour les hémochromatoses chez l’adulte il est donc proposé une approche pragmatique avec des investigations paracliniques guidées par le contexte, l’anamnèse et l’examen clinique. Pour les hémochromatoses survenant chez l’enfant au vu de la fréquence et du caractère parfois asymptomatique de l’atteinte cardiaque, une exploration systématique est recommandée au diagnostic, puis une surveillance adaptée en fonction de l’atteinte initiale.
Les manifestations extra hépatiques doivent être évaluées cliniquement chez les patients avec une hémochromatose. Cela concerne l’atteinte osseuse (arthropathie, ostéoporose), et endocrinienne (diabète, dysfonction érectile, perte de libido, aménorrhée).
Les patients avec une surcharge en fer sévère doivent être évalués sur le plan cardiaque par un électrocardiogramme et une échographie cardiaque. Si des signes cliniques cardiologiques sont associés, une IRM cardiaque avec quantification de la charge en fer doit être proposée.
Les patients avec une hémochromatose survenant avant l’âge adulte doivent systématiquement être évalués sur le plan cardiaque par un électrocardiogramme, une échographie et une IRM cardiaque.
Quels patients doivent être inscrits dans un programme de dépistage du Carcinome hépatocellulaire et à quelle fréquence ?
Le cancer primitif du foie, principalement le carcinome hépatocellulaire (CHC), est la première cause de mortalité des patients avec une hémochromatose HFE (8, 9). Le risque pour un patient homozygote HFE p.C282Y serait d’approximativement 10 fois celui de la population générale. Comme dans la plupart des maladies chroniques du foie le CHC survient chez un patient cirrhotique, chez qui doit donc être mis en place un dépistage. Cependant quelques cas ont été décrits chez des patients avec une fibrose avancée, mais aussi parfois avec une fibrose minime, voire absente. Dans les recommandations de l’EASL pour le CHC il est proposé de discuter la mise en place du dépistage chez les patients ayant un stade de fibrose F3 en fonction de l’évaluation du risque individuel. Bien que peu de données soient disponibles dans le contexte spécifique de l’hémochromatose, il semble approprié de suivre la même approche. Les risques identifiés de CHC chez un patient avec une hémochromatose sont un âge plus élevé, une surcharge en fer plus importante, la durée d’exposition à la surcharge en fer, le sexe masculin, la présence d’un diabète, la consommation d’alcool ou de tabac, et la présence d’une hépatite virale chronique. Les autres facteurs de risques généraux de carcinomes hépatocellulaires tels que la stéatopathie métabolique doivent aussi être pris en compte pour l’évaluation de ce risque.
La régression de la fibrose après traitement a été décrite, y compris chez des patients avec une cirrhose initiale au diagnostic, dans ces situations le risque de CHC est significativement réduit. Il n’existe cependant pas de critère permettant de guider la temporalité et les moyens de réévaluation de la fibrose, et donc de critère permettant d’interrompre le dépistage du CHC.
En l’absence de donnée précisant l’histoire naturelle du CHC dans l’hémochromatose, elle est considérée comme similaire aux autres maladies chroniques du foie. Un intervalle de 6 mois pour les examens de dépistages est donc conseillé. Il peut être associé au dosage de l’alpha fœto-protéine. Le but du dépistage étant d’améliorer la survie des patients, le dépistage n’est pertinent que chez les patients qui pourraient bénéficier d’un éventuel traitement de ce CHC.
Les patients ayant une hémochromatose et une cirrhose doivent bénéficier d’un programme de dépistage du carcinome hépatocellulaire tous les 6 mois indépendamment de la normalisation de la surcharge en fer.
Le dépistage, tous les 6 mois, peut être discuté chez les patients ayant une fibrose avancée (F3).
Le dépistage doit être effectué par échographie, le dosage de l’alpha fœto protéine peut y être associé. En cas de limitation technique de l’échographie, l’IRM ou le scanner peuvent être proposés.
Traitement et suivi
Quel est le traitement de première ligne de l’hémochromatose ?
Le traitement standard de l’hémochromatose est la saignée thérapeutique qui réduit la surcharge en fer en mobilisant le fer pour l’érythropoïèse. Ce traitement permet de significativement améliorer la survie et la morbidité des patients s’il est initié avant l’apparition de la cirrhose ou du diabète. Les données disponibles montrent que les saignées améliorent l’asthénie, les douleurs articulaires, l’atteinte cardiaque et l’atteinte hépatique.
Le traitement comporte deux phases, une phase initiale d’induction pour normaliser le stock en fer, puis une phase de maintenance visant à éviter la ré-accumulation. Bien que présentant certaines contraintes, les saignées sont généralement bien supportées et peu coûteuses. Les patients présentant une hémochromatose peuvent être donneurs de sang.
L’apparition d’une carence en fer, ou d’une réduction inexpliquée du besoin de saignée doit faire rechercher une cause sous-jacente de saignement occulte.
Une étude récente suggère que l’érythrocytaphérèse pourrait améliorer la fatigue et les paramètres biologiques du métabolisme du fer. Une étude randomisée de petite taille suggère que cela permettrait une normalisation plus rapide du stock en fer et un moindre nombre de procédures que les saignées avec une bonne tolérance. Cependant l’évaluation en termes de coût-efficacité dépend des techniques et de sa disponibilité.
Les patients diagnostiqués précocement sans surcharge en fer constituée peuvent être encouragés à être donneurs de sang.
Les patients ayant une hémochromatose avec une surcharge en fer doivent avoir un traitement déplétif du fer.
Le traitement de première intention est la saignée. L’érythrocytaphérèse peut être proposée en phase d’induction si elle est disponible.
Quand un traitement de deuxième ligne doit être discuté ?
Lorsque les saignées ne sont pas possibles, ou à un rythme insuffisant pour normaliser le stock en fer, un traitement chélateur peut être envisagé. Cela peut être le cas en l’absence d’abord veineux, de phobie des aiguilles, d’anémie concomitante (dont le bilan étiologique doit être fait, car l’hémochromatose n’engendre pas d’anémie), ou d’atteinte cardiaque sévère.
Des traitements par deferoxamine (perfusion), et hors autorisation de mise sur le marché par deferiprone (oral) ou deferasirox (oral) ont été décrits. Au vu des faibles données disponibles, ils sont à réserver aux centres en ayant l’expérience clinique. Le traitement par deferasirox est celui qui a fait l’objet de plus d’études dans le cadre d’un développement en phase I et II. Les résultats suggèrent une dose initiale de 10 mg/kg/j, à adapter en fonction de la réponse et de la tolérance.
Dans les formes juvéniles d’hémochromatose les saignées sont efficaces et bien tolérées, cependant la présence d’atteinte clinique sévère incite à envisager un traitement combiné avec un chélateur dans la phase initiale de normalisation du stock en fer. Le traitement de maintenance est assuré par la suite par saignée ou chélateur en fonction de l’évolution et de la tolérance. En situation de surcharge massive avec atteinte cardiaque sévère une association de chélateur a été décrite dans plusieurs cas cliniques avec des résultats intéressants.
Les traitements chélateurs sont contre indiqués pendant la grossesse. Leur posologie doit être adaptée à la fonction rénale. Les effets indésirables spécifiques à chaque traitement doivent être évalués et les patients surveillés de manière régulière.
En réduisant l’absorption du fer non héminique, les inhibiteurs de la pompe à protons peuvent être une aide utile chez certains patients. En effet une étude randomisée a montré que les inhibiteurs de la pompe à protons réduisaient de moitié le besoin en saignées des patients avec une hémochromatose (10). Bien qu’insuffisants de manière isolée ils peuvent être une aide ponctuelle dans la gestion de certains patients compliqués.
Lorsque les saignées ne sont pas réalisables, un traitement chélateur doit être débuté en évaluant la balance bénéfice/risque.
Le deferasirox est le chélateur qui bénéficie du plus de données et a montré son efficacité pour réduire la charge en fer. Les données restent cependant limitées, et il n’a pas d’autorisation de mise sur le marché.
Quels sont les objectifs du traitement ?
Les différentes sociétés savantes suggèrent des objectifs similaires, mais non strictement égaux. Tous sont basés sur la ferritine, avec des seuils qui peuvent être variables en fonction de la phase et des recommandations, en notant que finalement très peu de données objectives sont disponibles pour évaluer ces objectifs. En se basant sur la littérature, il a été proposé de façon empirique le seuil de 50 µg/L à la phase d’induction puis un seuil de 50 à 100 µg/L lors de la phase d’entretien.
Le volume et la fréquence des saignées pour atteindre ces objectifs sont de 400 à 500 ml tous les sept ou quinze jours en fonction du poids pendant la phase d’induction, puis tous les 1 à 4 mois lors de la phase d’entretien. Le taux d’accumulation du fer chez les patients avec hémochromatose varie grandement, avec une estimation d’une majoration de 100 µg/L par an en l’absence de traitement.
Le taux d’hémoglobine doit être surveillé au cours du traitement d’induction et d’entretien. Le traitement doit être diminué en cas d’hémoglobine <12 g/dl, et interrompu en cas d’hémoglobine <11 g/dl.
La ferritine doit être surveillée pour s’assurer de l’obtention de l’objectif du traitement sans traitement excessif. Le rythme de la surveillance est mensuel en phase d’induction, puis quand la ferritine est inférieure à 200 µg/L, toutes les unes à deux saignées. Pendant la phase d’entretien, elle doit être surveillée tous les 6 mois pour adapter le traitement. Un changement inattendu de la ferritine au cours du traitement d’entretien doit être exploré, car une fluctuation significative n’est pas habituelle dans l’hémochromatose.
La question de l’intérêt de la surveillance du coefficient de saturation de la transferrine au cours du traitement n’est pas résolue. La saturation peut rester élevée chez des patients dont la ferritine est dans l’objectif thérapeutique, et des données rétrospectives suggèrent que cela pourrait être associé à une moindre qualité de vie à long terme. Il n’y a cependant pas de donnée permettant de donner un objectif de traitement associé à une amélioration clinique.
Il est recommandé de vérifier périodiquement les taux de folates et de vitamines B12, en particulier en cas de saignée très fréquente. Si nécessaire une supplémentation peut être proposée.
Le traitement par saignée comporte une phase d’induction et une phase d’entretien.
Pendant la phase d’induction les saignées sont effectuées toutes les unes ou deux semaines avec un objectif de ferritine de 50 µg/L.
Pendant la phase d’entretien, la ferritine doit être maintenue entre 50 et 100 µg/L avec une certaine flexibilité.
Grossesse
Aucune étude ne s’est spécifiquement intéressée à l’évolution maternelle et fœtale au cours de l’hémochromatose. Considérant l’histoire naturelle de la maladie, il est peu probable que l’interruption des saignées le temps de la grossesse affecte significativement le devenir à long terme. Une grossesse normale est fortement consommatrice de fer, et la carence en fer est un facteur de risque maternel et fœtal qui doit être évité. Il est recommandé d’éviter la carence en fer avant et pendant la grossesse. Chez une patiente envisageant une grossesse, le traitement peut être allégé avec un objectif de ferritine >45 µg/L. La cirrhose et l’hypertension portale, si présentes, doivent être prises en charge dans le contexte de grossesse de façon similaire aux autres maladies chroniques du foie.
Chez une patiente envisageant une grossesse la carence en fer doit être évitée.
Chez une patiente avec une hémochromatose et une surcharge modérée ou faible sans signe de maladie du foie avancée, la décision concernant le traitement peut être individualisée, mais les saignées peuvent être arrêtées pendant la durée de la grossesse chez la plupart des patientes.
Quelles sont les recommandations diététiques dans l’hémochromatose ?
Il est fréquent que les patients s’interrogent sur les éventuelles restrictions alimentaires à adopter, et puissent parfois adopter des modifications alimentaires inappropriées. De façon générale les patients ayant une hémochromatose doivent éviter les supplémentations en fer ou en vitamine C ainsi qu’une consommation d’alcool excessive.
La vitamine C est une amplificatrice puissante de l’absorption du fer minéral, elle peut aussi jouer un rôle pro oxydant. Ses interactions avec le métabolisme du fer sont complexes, ainsi chez les patients ayant une thalassémie majeure et une surcharge en fer, la supplémentation en vitamine C est associée à une dégradation rapide de la fonction cardiaque. La consommation de fruits et légumes ne doit pas être limitée, en revanche la consommation de fruits ou jus de fruits riche en vitamine C doit se faire préférentiellement de façon isolée pour ne pas augmenter l’absorption de fer par ailleurs.
Le fer lié à l’hémoglobine est absorbé de façon efficace et sa biodisponibilité est peu affectée par la composition alimentaire. Les patients ayant une hémochromatose ont une absorption significativement supérieure du fer minéral et du fer lié à l’hémoglobine y compris en cas de surcharge en fer. Pour limiter l’absorption de fer, la consommation de viande rouge peut être limitée.
Certains aliments (tels que le thé noir ou le curcuma) peuvent limiter l’absorption du fer. Les modifications alimentaires peuvent aider à réduire l’accumulation de fer ou le nombre de saignées requises, mais il n’y a pas de donnée montrant un bénéfice clinique significatif.
La consommation excessive d’alcool majore le niveau de surcharge en fer et accélère la progression de la fibrose hépatique vers la cirrhose, elle est associée à une augmentation de la mortalité de cause hépatique et de l’incidence du CHC. L’impact d’une consommation modérée est moins clairement établi dans l’hémochromatose. Cependant de façon générale une consommation modérée (10-20 g/jours) d’alcool est associée à une surmortalité liée au foie ou par cancer.
La consommation d’alcool doit donc toujours être évaluée chez un patient ayant une surcharge en fer. Il doit être conseillé au patient d’éviter la consommation d’alcool ou de façon réduite, en fonction des seuils, considérée comme à risque tout en prenant en compte les autres facteurs de risque hépatique potentiels. En présence d’une surcharge en fer ou d’une atteinte hépatique, la consommation d’alcool devrait être exceptionnelle.
Les infections à Vibrio vulnificus, qui est une bactérie Gram négatif présente dans les fruits de mer, peuvent être particulièrement graves chez les patients présentant une surcharge en fer. La mortalité est supérieure à 50 % en cas de septicémie, cette virulence s’expliquant par la très forte affinité de cette bactérie pour le fer. Chez les patients avec un stock en fer normal ce risque est atténué.
Les conseils alimentaires ne doivent pas être substitués au traitement déplétif en fer.Les supplémentations en fer ou en vitamine C doivent être évitées.
La consommation de viande rouge devrait être modérée.
La consommation d’alcool doit être limitée aux seuils considérés comme à risque. Chez les patients ayant une surcharge en fer ou une atteinte hépatique la consommation doit être évitée.
Les fruit et jus de fruits, en particulier agrumes, riches en vitamines C doivent être consommés avec modération et de préférence sans association aux autres aliments.
Chez les patients ayant une hémochromatose et une surcharge en fer la consommation de fruits de mer peut être associée à une infection grave à Vibrio vulfinicus.
Références
- Girelli D, Busti F, Brissot P, Cabantchik I, Muckenthaler MU, Porto Hemochromatosis classification: update and recommendations by the BIOIRON Society. Blood. 19 mai 2022;139(20):3018-29.
- Porto G, Brissot P, Swinkels DW, Zoller H, Kamarainen O, Patton S, et EMQN best practice guidelines for the molecular genetic diagnosis of hereditary hemochromatosis (HH). Eur J Hum Genet. avr 2016;24(4):479-95.
- Powell LW, Seckington RC, Deugnier Haemochromatosis. The Lancet. août 2016;388(10045):706-16.
- Brissot P, Pietrangelo A, Adams PC, de Graaff B, McLaren CE, Loréal Haemochromatosis. Nat Rev Dis Primer. 5 avr 2018;4:18016.
- Sandhu K, Flintoff K, Chatfield MD, Dixon JL, Ramm LE, Ramm GA, et Phenotypic analysis of hemochromatosis subtypes reveals variations in severity of iron overload and clinical disease. Blood. 5 juill 2018;132(1):101-10.
- Pilling LC, Tamosauskaite J, Jones G, Wood AR, Jones L, Kuo CL, et Common conditions associated with hereditary haemochromatosis genetic variants: cohort study in UK Biobank. BMJ. 16 janv 2019;364:k5222.
- Benito-Lozano J, Arias-Merino G, Gómez-Martínez M, Arconada-López B, Ruiz-García B, Posada de la Paz M, et Psychosocial impact at the time of a rare disease diagnosis. PloS One. 2023;18(7):e0288875.
- Bardou-Jacquet E, Morcet J, Manet G, Lainé F, Perrin M, Jouanolle AM, et Decreased cardiovascular and extrahepatic cancer-related mortality in treated patients with mild HFE hemochromatosis. J Hepatol. mars 2015;62(3):682-9.
- Atkins JL, Pilling LC, Masoli JAH, Kuo CL, Shearman JD, Adams PC, et Association of Hemochromatosis HFE p.C282Y Homozygosity With Hepatic Malignancy. JAMA. 24 nov 2020;324(20):2048-57.
- Vanclooster A, van Deursen C, Jaspers R, Cassiman D, Koek Proton Pump Inhibitors Decrease Phlebotomy Need in HFE Hemochromatosis: Double-Blind Randomized Placebo-Controlled Trial. Gastroenterology. sept 2017;153(3):678-680.e2.