Objectifs pédagogiques
- Connaître les molécules disponibles, leurs indications et leurs modalités de prescription
- Connaître l’efficacité des anti-IL-23 et leur profil de tolérance
- Connaître leur positionnement dans la stratégie thérapeutique
Toute reproduction ou réécriture, totale ou partielle, sans l’accord préalable écrit de la FMC HGE est interdite.
Abbvie-Janssen-Takeda-Celltrion-Amgen-Lilly-Pfizer-Biocon
Anti-IL23/Maladie de Crohn/Rectocolite hémorragique.
Autorisation de Mise sur le Marché : AMM
Anti interleukine : anti-IL
Anti-TNF : anti-Tumor Necrosis Factor
CDAI: Crohn Disease Activity Index
European Medicines Agency: EMA
FDA: Food and Drug Administration
Immunoglobuline : Ig
Intra Veineux : IV
Janus Kinase : JAK
Maladie de Crohn : MC
MICI : Maladies Inflammatoires Chroniques de l’Intestin Monoclonal
Anti Bodies: mAb
Natural Killer: NK
Nouvelles Thérapies Avancées : NTA
Patient Reported Outcome : PRO
Rectocolite hémorragique : RCH Semaine: S
Signal Transducers And Transcription: STAT
Simple Endoscopic Score-Crohn Disease: SES-CD
Sous Cutané : SC
Tyrosine Kinase : TyK
Au cours de ces dernières années de nouvelles classes thérapeutiques sont apparues pour le traitement des Maladies Inflammatoires Chroniques de l’Intestin (MICI), permettant d’enrichir les possibilités de prise en charge avec néanmoins un taux d’efficacité prolongée qui stagne autour de 50 % et qui constitue encore à l’heure actuelle un « plafond de verre » (1). Parmi les Nouvelles Thérapies Avancées (NTA) et après l’arrivée des anti-TNF depuis une vingtaine d’années (2), les anti-IL-23 viennent enrichir l’arsenal thérapeutique avec un rapport bénéfice/risque amélioré (3). La compréhension exacte de leurs mécanismes d’action reste encore à préciser, ainsi que leur positionnement parmi les différentes lignes thérapeutiques disponibles (4, 5).
L’objet de cette mise au point est de clarifier le mode d’action des anti-IL-23, de rendre compte des résultats d’efficacité et de tolérance à partir des études disponibles, puis d’évoquer leur positionnement dans les différentes séquences thérapeutiques avec leurs perspectives d’utilisation.
L’IL-12 et IL-23 sont des cytokines hétérodimériques à potentiel pro-inflammmatoire qui partagent la même sous unité p40. L’IL-12 est formée des sous unités p40 et p35 qui se fixent sur leur récepteur spécifique au niveau de la membrane cellulaire (composé de 2 sous unités IL-12 R b1 et b2) tandis que l’IL-23 est composée des sous unités p40 et p19 (6). La liaison d’IL-23 avec son récepteur spécifique qui est un hétérodimère transmembranaire composé de la sous unité IL-12 R b1 et IL-23 R, conduit à l’activation d’une voie de signalisation intracellulaire incluant les Janus kinases (JAK) 2, TYK2 et le facteur de transcription STAT 3 (Fig. 1) tandis que l’IL-12 active la voie intra cellulaire STAT 4.
Ces deux interleukines ont des similitudes structurelles, mais leurs rôles respectifs dans les réponses immunitaires sont différents. L’IL-12 stimule principalement la différenciation des lymphocytes T naïfs vers des lymphocytes TCD4+ Th1 qui produisent de l’IL-2 et de l’interféron-y. L’IL-23 stabilise la différenciation des lymphocytes TCD4+ Th 17 qui produisent de l’IL-17A/IL-17 F, du TNFa, de l’IL-6/IL-22 et des facteurs de stimulation des monocytes granulocytaires (7, 8). Les macrophages, les cellules dendritiques et les polynucléaires neutrophiles sont les principales sources de production d’IL-23 (9, 10, 11). Les principales cellules immunitaires cibles de l’IL-23 sont les lymphocytes T Th17, les ILC3 (cellules lymphoïdes innées) et les lymphocytes Tgô.
Fig. 1 : D’après Vuyyuru SK et al. (5). Voies de signalisation intra cellulaires via IL-12 et IL-23
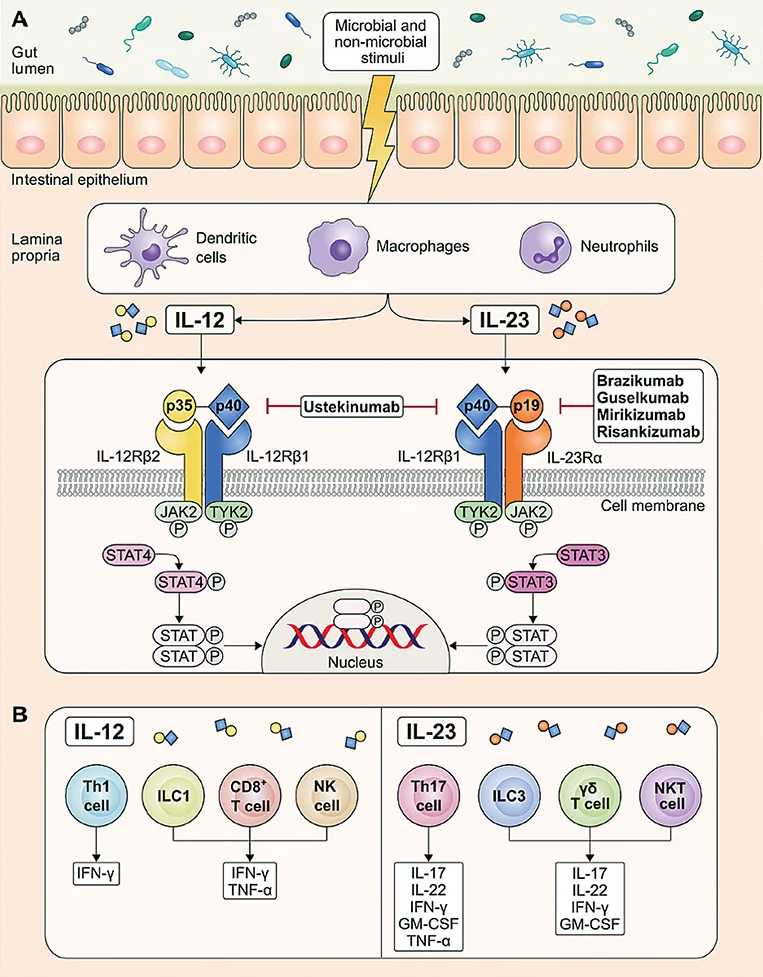
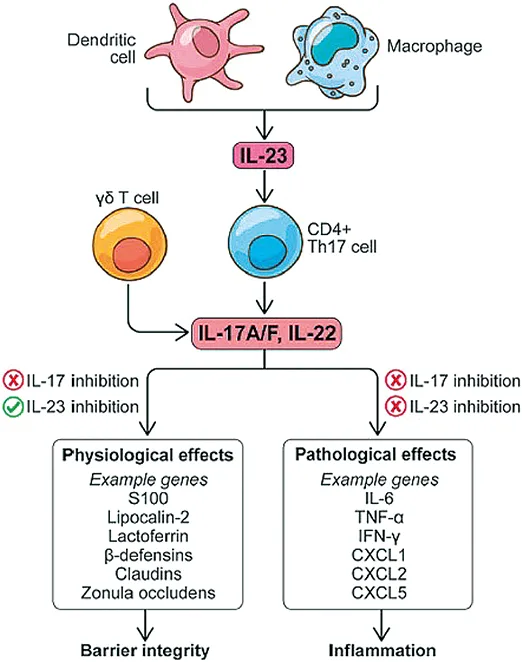
L’IL-12 et l’IL-23 sont deux cytokines présentes à l’état normal, en particulier au niveau de l’iléon distal. On observe une élévation de leurs concentrations en cas d’infection ou d’inflammation. Il a été montré que l’inhibition d’IL-12 via des anticorps neutralisant anti p40 ou le blocage de l’expression de son gène, protégeait de l’apparition de pathologies auto-immunes induites (12). En outre, le blocage d’IL-23 p19 était plus efficace comparé au blocage d’IL-12 p40 suggérant que la voie de l’IL-23 était la voie prédominante dans l’inflammation chronique. Il existe des polymorphismes génétiques d’IL-23 responsables de susceptibilités ou de protections vis-à-vis de l’apparition d’une MICI. Il a même été évoqué un rôle prédominant de l’IL-23 et de l’IL-17 à l’origine des MICI. L’IL-23 joue également un rôle dans le maintien de l’intégrité de la muqueuse intestinale et des mécanismes de cicatrisation via l’IL-17 et l’IL-22. Maxwell et al (13) ont montré dans un modèle expérimental sur la souris, que l’inhibition de l’IL-17 pouvait être responsable de lésions de la barrière intestinale. Cela contribue certainement à expliquer l’inefficacité des antagonistes de l’IL-17 comme le sécukinumab, l’ixekinumab ou le brodalumab qui peuvent même aggraver une poussée de MICI ou déclencher une MICI inaugurale. En résumé, l’IL-23 via l’induction de l’IL-22 par les lymphocytes et l’activation de la voie STAT 3, joue un rôle majeur dans le contrôle de la cicatrisation muqueuse au cours des MICI (13,14,15-Fig. 2).
Une activation inappropriée de la voie de l’IL-23 et des concentrations élevées d’IL-17 ont aussi été rapportées au cours de plusieurs maladies dysimmunitaires inflammatoires chroniques (psoriasis, rhumatismes inflammatoires et MICI) (5).
L’ustékinumab qui cible la sous unité p40 commune à l’IL-12 et l’IL-23 est le premier anticorps monoclonal de cette classe thérapeutique approuvée initialement dans le traitement de la MC puis de la RCH (16,17). Des anticorps dirigés sélectivement contre la sous unité p19 de l’IL 23 ont été développés et approuvés ces dernières années dans le traitement du psoriasis (risankizumab, guselkumab, tildrakizumab), du rhumatisme psoriasique (risankizumab et guselkumab), et de façon plus récente, de la MC et de la RCH (risankizumab, guselkumab, mirikizumab) (18, 19, 20, 21). D’un point de vue structural ces trois immunoglobulines ont de grandes similarités avec néanmoins des différences en particulier au niveau de leur domaines Fc. Il en va de même d’un point de vue pharmacocinétique. Plus récemment, une analyse transcriptomique in vitro de biopsies intestinales de patients atteints de MICI, concomitante à un test de co-culture in vitro de monocytes CD64+ (activés pour produire de l’IL23) et d’une lignée cellulaire rapporteur d’IL-23 (sensible à l’IL-23 biologiquement active), a démontré une puissance accrue in vitro du guselkumab par rapport au risankizumab pour inhiber la signalisation de l’IL-23. Le guselkumab, mais pas le risankizumab, est capable de se lier à la cellule CD64+ (FcgRI) via sa région Fc native, ce qui pourrait neutraliser l’IL-23 produite localement par les cellules CD64+ à proximité des cellules sensibles à l’IL-23. Cet effet pourrait potentiellement contribuer aux différences d’efficacité entre ces différents anticorps, ce qui reste néanmoins à confirmer (22, 23).
Fig. 2 : D’après Bourgonje et al (23). Rôles respectifs d’IL -23, IL- 22 et IL-17 dans l’homéostasie et les effets pathologiques induits au cours des MICI
Au total, trois biothérapies anti-IL-23 ont été évaluées dans des programmes de développement dans les MICI (risankizumab, mirikizumab et guselkumab) (19, 21, 24, 25).
Le risankizumab est un anticorps monoclonal (mAb) IgG1 humanisé, avec un domaine Fc muté (LALA) produit par des cellules ovariennes de hamster chinois par la technique de l’ADN recombinant. Il a d’abord été évalué dans un essai randomisé en double aveugle de phase 2 chez 121 patients présentant une MC modérée à sévère. Une randomisation en trois bras équilibrés a été effectuée afin d’administrer aux patients inclus un traitement par risankizumab IV à la dose de 200 ou 600 mg versus placebo, aux semaines S0, S4 et S8. Une majorité de malades avaient déjà été exposés à au moins un anti-TNF au moment de l’inclusion dans cette étude (19). La réponse était évaluée à partir d’un score CDAI < 150 à la semaine S12. Les résultats étaient positifs pour ce qui concerne les groupes risankizumab. En revanche, l’efficacité paraissait supérieure à la dose de 600 mg, avec un delta d’efficacité de 36,6 % versus 24,4 % pour la dose à 200 mg en comparaison du placebo. Concernant les objectifs secondaires (CDAI< 150, réduction du score CDAI> 50 %, et la rémission endoscopique – CDEIS≤ 4 ou CDEIS≤ 2 – pour les patients avec une atteinte iléale isolée) à S12, la dose de 600 mg était statistiquement supérieure au placebo alors que celle de 200 mg n’apparaissait supérieure au placebo que pour l’obtention d’une rémission endoscopique.
Par la suite, le programme de développement de la molécule a consisté en une étude de phase 3 d’induction (études ADVANCE et MOTIVATE) (26) avec une étude de maintenance (FORTIFY) (27), qui ont confirmé les résultats initiaux.
L’étude ADVANCE (26-Fig. 3) a permis d’inclure 931 patients qui étaient préalablement en échec d’un traitement par biothérapie (58 %). L’étude MOTIVATE a quant à elle inclus 618 patients tous en échec d’une biothérapie précédente. Dans les populations en intention de traiter, 110 (22 %) des 491 patients dans ADVANCE et 109 (19 %) des 569 patients dans MOTIVATE avaient eu une réponse inadéquate ou une intolérance à l’ustékinumab. Les deux études étaient randomisées avec un schéma d’induction à 600 ou 1200 mg IV versus placebo. Les objectifs de ces deux études étaient la rémission clinique et la réponse endoscopique à la semaine S12. Dans ces deux essais, 30 % et 53 % des patients avaient été considérés en échec après au moins deux traitements biologiques et environ 20 % d’entre eux (dans les deux essais) avait déjà été traités préalablement par de l’ustékinumab. Les résultats étaient significativement positifs pour les patients inclus dans les bras risankizumab avec une atteinte de l’objectif principal en termes de rémission clinique et de réponse endoscopique à S12. Un plus grand pourcentage de réponse était observé dans le groupe des patients naïfs de biologiques. Dans un second temps, les répondeurs cliniques à S12 étaient randomisés en trois bras pour un traitement de maintenance en double aveugle avec du risankizumab sous-cutané à la dose de 180 mg ou 360 mg toutes les huit semaines versus placebo. Les patients traités au long cours par risankizumab ont atteint de meilleurs taux de réponse, de rémission clinique (CDAI ou PRO) et de réponse endoscopique à S52 (27).
Fig. 3 : D’après G. D’Haens et al. Étude d’induction ADVANCE (26). Co-critères primaires d’évaluation à S12
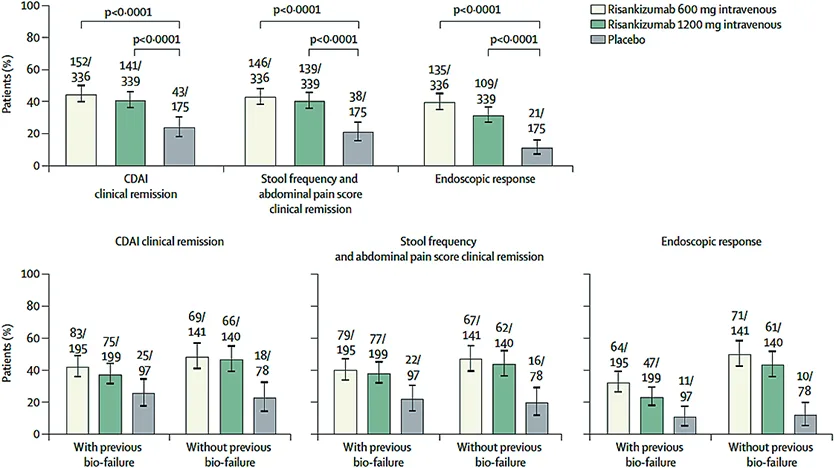
Fig. 4 : D’après M. Ferrante et al. Étude FORTIFY (27). Co-critères primaires d’évaluation à S52
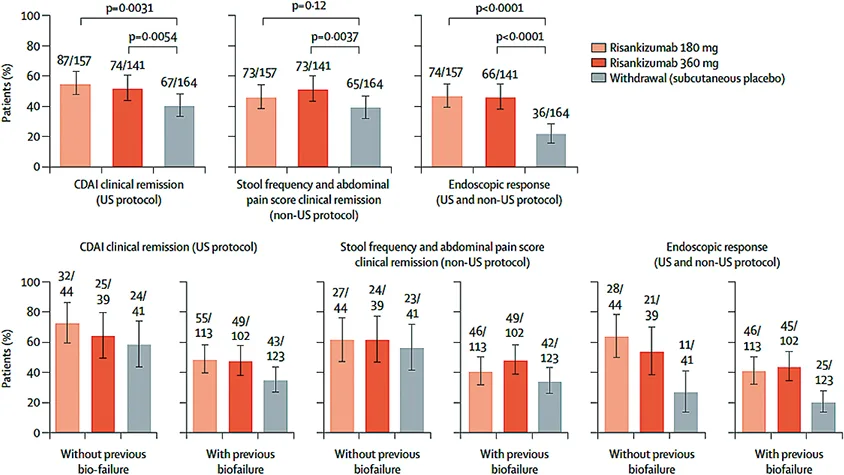
Les résultats de la semaine 52 pour les principaux critères d’évaluation secondaires ont montré également des taux d’efficacité plus élevés pour les critères objectifs de rémission endoscopique et d’endoscopie sans ulcère (c’est-à-dire l’absence d’ulcération), les critères composites de rémission clinique et de réponse endoscopique, et la profondeur de la rémission dans les groupes de traitement par risankizumab par rapport au groupe d’arrêt (placebo sous-cutané). L’effet thérapeutique était en général plus important avec la dose de 360 mg par rapport à la dose de 180 mg. De plus, un effet thérapeutique a été observé chez les patients avec et sans échec biologique antérieur, avec néanmoins un taux de réponses plus élevé chez les patients bio-naifs.
Pour les patients qui n’avaient pas répondu à S12 sous risankizumab, une nouvelle induction était possible pour une période supplémentaire de 12 semaines par risankizumab en injection sous-cutanée à la posologie de 180 mg ou 360 mg dans l’étude FORTIFY (27-Fig. 4). Parmi ces patients, la rémission clinique a été de 53,3 % et 66,7 %, aux doses 180 mg et 360 mg, respectivement.
Dans une première étude de vraie vie française, multicentrique, rétrospective, Fumery et al. (28), ont montré dans une population de MC réfractaire (tous les patients avaient reçu au moins trois biologiques), que le traitement par risankizumab a permis l’obtention d’une rémission clinique sans corticoïdes à un an chez 46 % des patients avec un taux de persistance de 79 %. Le profil de sécurité est apparu identique à celui rapporté précédemment dans la littérature.
Le risankizumab est le premier anti- IL-23 à avoir obtenu une AMM dans la maladie de Crohn (29). Son périmètre de remboursement est encore en attente. Il dispose également d’une AMM dans le psoriasis et le rhumatisme psoriasique.
Le mirikizumab est un anticorps monoclonal (mAb) IgG4 humanisé avec un domaine Fc muté (FALA) qui inhibe l’IL-23 en se liant à l’épitope de sa sous-unité p19.
L’étude SERENITY de phase 2, en double aveugle, a permis d’inclure 191 patients atteints de formes modérées à sévères de MC (21). Il s’agissait d’une étude randomisée avec quatre bras (2 :1 :1 :2) : un bras placebo IV, et trois bras avec le mirikizumab à 200, 600 et 1000 mg en IV toutes les quatre semaines. L’objectif principal était l’obtention d’une réponse endoscopique avec une réduction de 50 % du score SES-CD à S12. Les patients en réponse clinique (amélioration supérieure ou égale à un point de ce score endoscopique) à S12, étaient re-randomisés pour continuer soit le traitement IV à la dose d’induction, soit avec une dose de 300 mg en injection sous-cutanée toutes les quatre semaines. Les patients en maintenance recevaient à chaque fois 2 injections, une IV et une sous-cutanée afin de conserver le double aveugle. L’efficacité du mirikizumab était supérieure au placebo à S12, tant sur le plan endoscopique que clinique (évaluation des PRO), pour les dosages à 600 et 1000 mg uniquement. La réponse endoscopique paraissait meilleure pour les patients traités à la dose de 1000 mg, alors que la réponse clinique était optimale à la dose de 600 mg. Dans la phase de maintenance, la rémission endoscopique à S52 variait entre 19,5 % pour la voie IV et 32,6 % pour la forme sous-cutanée. La réponse histologique, évaluée sur différents scores, apparaissait meilleure et significativement supérieure au placebo à une dose de 1000 mg de mirikizumab. À S52, en tenant compte des petits effectifs étudiés, plus de 58 % des patients, avec à l’inclusion une activité histologique marquée, ont eu une réponse histologique versus une rémission histologique qui variait de 19,5 à 32,6 % en fonction des bras traités.
VIVID-1 est un essai de phase 3, multicentrique, randomisé (3 bras), en double aveugle, « treat through » (absence de re-randomisation après l’induction avec des patients qui gardent leur traitement jusqu’à la fin d’étude), contrôlé versus placebo et un traitement actif (ustekinumab), évaluant l’efficacité et la tolérance du mirikizumab chez des patients atteints de MC active, modérée à sévère.
Les résultats ont été présentés au congrès de la Digestive Disease Week (DDW) en 2024 (30). Les objectifs étaient de préciser l’efficacité et la tolérance du mirikizumab en comparaison au placebo jusqu’à S52 et d’évaluer à partir de critères secondaires, l’efficacité du mirikizumab par rapport à l’ustékinumab. Au total, 1 150 patients ont été randomisés (6:3:2) entre le mirikizumab (900 mg par voie IV à S0, S4 et S8, puis 300 mg SC mensuel), l’ustekinumab (6 mg/ kg IV à S0, puis 90 mg SC toutes les 8 semaines) ou le placebo. Les patients non répondeurs dans le groupe placebo étaient traités dans un second temps dans un 4e bras d’induction et de maintenance avec du mirikizumab.
À S12, la réponse endoscopique était observée chez 32,5 % des patients du groupe mirikizumab vs. 12,6 % dans le groupe placebo (p< 0,000001) ; les taux de rémission endoscopique étaient respectivement de 17,6 % vs. 7,0 % (p < 0,0002).
Les résultats à S52 ont révélé qu’une proportion plus élevée des patients du groupe mirikizumab avaient atteint les co-critères d’évaluation primaires par rapport au groupe placebo. Au total, 48,4 % dans le groupe mirikizumab vs. 9,0 % dans le groupe placebo ont obtenu une réponse endoscopique (P< 0,000001). De même, une proportion plus élevée de patients a obtenu une rémission clinique évaluée sur le CDAI : 54,1 % dans le groupe mirikizumab vs. 19,6 % dans le groupe placebo (P< 0,000001).
Dans l’ensemble, 38 % des patients traités par mirikizumab contre 9 % dans le groupe placebo ont atteint un critère composite de réponse clinique (PRO) à S12 et de réponse endoscopique évaluée à partir du SES-CD à S52 (P< 0,000001).
L’efficacité clinique du mirikizumab à S52 était indépendante de l’exposition antérieure ou de l’échec à une précédente biothérapie. Le mirikizumab était non inférieur à l’ustekinumab pour la rémission clinique et la réponse endoscopique à S52.
La tolérance des deux molécules était comparable.
Le guselkumab est un anticorps monoclonal IgG1 totalement humanisé avec un domaine Fc sauvage non muté, qui se fixe sélectivement sur la sous unité p19 de l’IL-23 et dont l’efficacité a été évaluée au cours d’une étude de phase 2 randomisée en double aveugle, chez 309 patients atteints de maladie de MC modérée à sévère (GALAXI 1) (24).
La randomisation a été faite en quatre bras (1:1:1:1) avec 3 doses IV de guselkumab toutes les quatre semaines (200 mg, 600 mg, 1200 mg) et un bras placebo. La randomisation intégrait également un bras de référence avec de l’ustékinumab à la dose de 6 mg/kg IV à S0, suivie d’une injection sous-cutanée à la dose de 90 mg à S8. Dans cette étude, environ 50 % des patients avaient déjà été traités ou avaient été considérés comme intolérants à un précédent traitement biologique. L’objectif principal était la diminution du CDAI à S12, observée de manière significative dans tous les groupes traités avec le guselkumab, comparativement au placebo. Il n’y avait cependant pas de corrélation évidente entre le niveau de réponse et la dose de guselkumab. Les résultats étaient également positifs pour les objectifs secondaires comme la diminution du CDAI <150 (53 % versus 16,4 % ; p< 005), la réponse endoscopique (diminution de 50 % du SES-CD par rapport au score de départ ou un SES-CD ≤ 2, 35,7 % versus 11,5 % ; p< 005), la réponse clinique (diminution du CDAI de 100 par rapport à l’évaluation initiale ou un score CDAI< 150, 65,9 % vs. 24,6 % ; p< 005), une amélioration des PRO2 et enfin, une amélioration des biomarqueurs, comme la CRP et la concentration de calprotectine fécale (de 47 % versus 6,6 % ; p < 0,05).
Dans le sous-groupe des patients préalablement traités par une biothérapie, 47,5 % des patients traités par guselkumab versus 10 % dans le groupe placebo ont obtenu une rémission clinique à S12.
Les patients répondeurs à S12 ont ensuite été traités en ouvert pendant 48 semaines par une injection SC toutes les :
La rémission clinique à S48 sous guselkumab variait de 57,4 % à 73 % et la réponse clinique, de 67,2 % à 84,1 %.
Actuellement des études de phase 3 sont en cours afin d’évaluer l’efficacité du guselkumab chez les patients atteints de maladie de Crohn modérée à sévère (GRAVITI/ NCT05197049), ainsi que chez les patients avec atteinte ano-périnéale (étude FUSION CD- NCT05347095).
Le mirikizumab a d’abord été étudié dans une étude de phase 2 concernant la sécurité et l’efficacité chez les patients atteints de RCH avec une forme modérée à sévère (31). Cet essai a inclus 249 patients avec une randomisation en 4 bras (1:1:1:1) : 50 mg, 200 mg, 600 mg et placebo, administrés en IV à S0, S4 et S8. Les patients considérés en réponse clinique (score de Mayo) étaient de nouveau randomisés à S12 pour un traitement de maintenance en injection sous-cutanée à la dose de 200 mg toutes les 4 ou 12 semaines, alors que les patients répondeurs sous placebo continuaient les injections toutes les quatre semaines. En termes de résultats, seule la dose de 200 mg de mirikizumab était statistiquement supérieure au placebo pour l’obtention d’une rémission clinique (22,6 % versus 4,8 %, p< 0,01) à S12. Néanmoins, les 2 doses évaluées à 50 et 200 mg étaient également significativement supérieures versus placebo pour l’induction de la réponse clinique à S12. En stratifiant les patients selon l’exposition préalable aux biothérapies, des différences significatives étaient observées (vs. placebo) uniquement chez les patients bio naïfs. Ainsi, seule la dose à 200 mg était supérieure au placebo pour l’induction de la réponse et de la rémission clinique, alors que les doses à 50 et 200 mg étaient toutes les deux supérieures versus placebo pour l’amélioration des scores endoscopiques.
À S52, 46,8 % et 37 % des patients qui avaient préalablement répondu à S12 et qui ont été traités avec une dose de 200 mg SC de mirikizumab respectivement toutes les 4 et 12 semaines, ont atteints une rémission clinique. L’élément original de cette étude était la possibilité d’une réinduction IV de 12 semaines supplémentaires à 600 ou 1000 mg chez les patients qui étaient en échec du traitement initial d’induction. Cela a fait l’objet d’un amendement per protocole. En termes de résultats et dans ce groupe de patients initialement en échec, la réinduction a permis une réponse clinique chez 50 % et 43,8 % respectivement aux doses de 600 et 1000 mg de mirikizumab. À S52, près de 69 % d’entre eux étaient en réponse clinique après un relais en traitement sous-cutané de maintenance à 200 mg toutes les quatre semaines.
Par la suite, le développement de la molécule a consisté en la réalisation de deux essais de phase 3, LUCENT 1 (induction) et LUCENT 2 (maintenance) consistant en un traitement par mirikizumab sur une période totale de 52 semaines (12 semaines d’induction + 40 semaines de maintenance) (32).
L’étude d’induction (LUCENT 1-Fig. 5) a permis l’inclusion de 1 281 patients adultes présentant une forme modérée à sévère de RCH. Ces patients avaient la particularité d’avoir une réponse insuffisante, une perte de réponse ou une intolérance aux corticoïdes, immunosuppresseurs, traitements biologiques ou au tofacitinib. La randomisation s’est faite en deux bras (3 : 1) avec 1 injection IV de mirikizumab 300 mg versus placebo à S0, S4 et S8. L’objectif principal était l’obtention d’une rémission clinique comme précédemment définie à S12. Les résultats montraient une efficacité supérieure du mirikizumab versus placebo à S12 (24,2 % versus 13,3 % ; p< 0,001). De la même façon la réponse clinique a S12 était supérieure au bras placebo (63,5 % versus 42,2 % ; P< 0,00001). La réponse était significativement meilleure versus placebo chez les patients naïfs de traitement biologique (70,1 % versus 50,3 % ; p < 0,001) comparativement à ceux déjà traités (54,6 % versus 29,7 % ; PR< 0,001). Le mirikizumab est apparu plus efficace pour ce qui concerne l’atteinte des objectifs secondaires y compris l’urgence intestinale (p< 0,001). La molécule était également supérieure au placebo pour le contrôle de la CRP (≤ ou > 6 mg/l – 59,3 % vs. 34,7 % ; p<0,001 %), la calprotectine fécale (≤ ou > 250 ug/g – 34,3 % vs. 20,1 % ; p<0,001) ainsi que pour l’obtention de la rémission histologique à S12 évaluée à partir du score de Geboes (29,3 % versus 15,6 % ; p< 0,001).
L’étude LUCENT-2 (32) (Fig. 6), essai randomisé multicentrique de 40 semaines en entretien, mené sur 367 centres dans 34 pays a évalué 544 patients sur les 551 patients ayant obtenu préalablement une réponse clinique à S12 (critère secondaire) avec le mirikizumab dans l’étude LUCENT-1. Une réduction progressive des corticoïdes était nécessaire à l’entrée dans LUCENT-2 pour les patients exposés. Ainsi, les malades en réponse clinique sous mirikizumab à S12 à l’issue de la phase d’induction (LUCENT 1), étaient de nouveau randomisés selon un ratio 2:1, en double aveugle pour un traitement de maintenance sous-cutané comparant du mirikizumab 200 mg versus placebo toutes les quatre semaines jusqu’à la semaine 52 (LUCENT 2). À l’issue de cette période de maintenance, près de 50 % des patients randomisés dans le groupe mirikizumab avaient atteint l’objectif principal concernant la rémission clinique versus 25,1 % des patients traités dans le groupe placebo (p< 0,001). La molécule est également apparue efficace dans l’évaluation des objectifs secondaires intégrant la fréquence des selles et les rectorragies. Près de 98 % des patients traités par mirikizumab en rémission clinique à S40 ne prenaient plus de corticoïdes. Le maintien de la rémission clinique était obtenu à S52 chez 63,6 % des patients traités par mirikizumab versus 36,9 % dans le groupe placebo. La rémission et/ou l’amélioration histologique à S52 ont pu être constatées respectivement chez 54,8 %/ 48,5 % parmi les patients traités par mirikizumab, versus 24,6%/25,7 % dans le groupe placebo (p< 0,001).
Fig. 5 : D’après G. D’Haens et al. Résultats de l’étude LUCENT 1 (32)
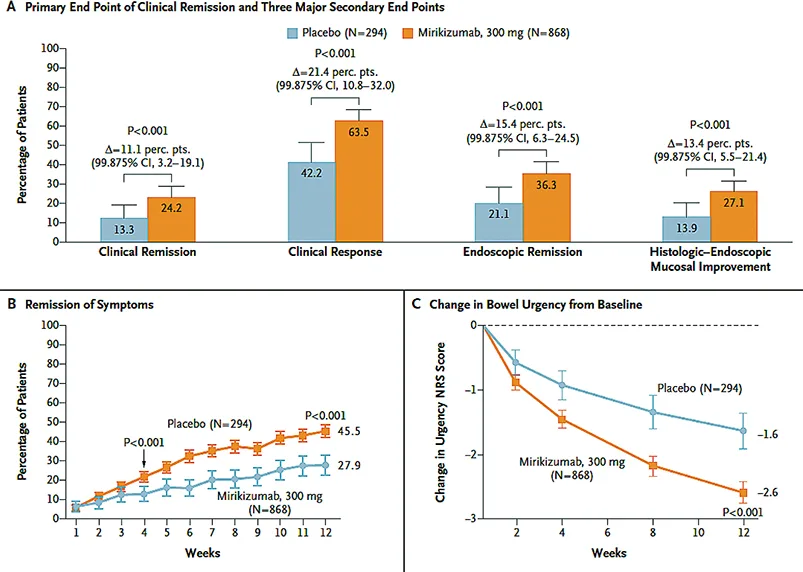
Fig. 6 : D’après G. D’Haens et al. Résultats de l’étude Lucent 2 (32)
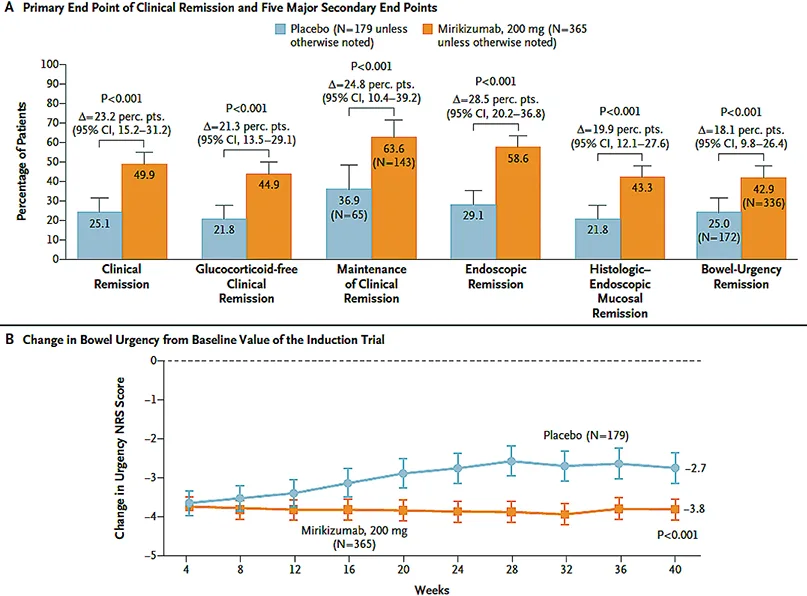
Le mirikizumab est le premier anti- IL-23 à avoir obtenu une AMM dans la RCH avec un périmètre de remboursement encore en attente pour la France (33).
L’étude de phase 2b QUASAR (34) a permis d’évaluer en double aveugle versus placebo l’efficacité du guselkumab en traitement d’induction chez 313 patients inclus pour une forme modérée à sévère de RCH. Les patients étaient randomisés de façon équilibrée en trois bras de traitement avec une perfusion de guselkumab à 200 ou 400 mg ou du placebo aux semaines S0, S4 et suivantes. L’objectif principal était la réponse clinique à S12. Le guselkumab était significativement plus efficace que le placebo pour la réponse clinique avec une atteinte également des objectifs secondaires concernant la rémission clinique, le contrôle des symptômes, l’amélioration endoscopique et un critère composite histo-endoscopique. Il n’a pas été constaté d’effet dose par rapport à la réponse. Parmi les patients inclus, 47,3 % d’entre eux avaient précédemment connus une réponse insuffisante ou une intolérance à deux précédents traitements par biothérapies ou tofacitinib, et environ la moitié des patients avaient eu une réponse insuffisante ou une intolérance vis-à-vis de plus de deux thérapies avancées. Le guselkumab s’est révélé également efficace pour le contrôle de la CRP et la diminution des concentrations de calprotectine fécale à S12 et dès S4.
Les données de maintenance de phase 3 (QUASAR) ont été récemment publiées (35). La rémission clinique à S44 a été obtenue par une proportion significativement plus élevée de patients traités par guselkumab SC, 200 mg toutes les 4 semaines (50 % [95 patients sur 190] ; différence de traitement ajustée 30 %, IC à 95 % 21-38 ; p<0,0001) et 100 mg toutes les 8 semaines (45 % [85 patients sur 188] ; différence de traitement ajustée 25 %, 16-34 ; p<0,0001) versus placebo (19 % [36 patients sur 190]).
L’étude exploratoire de phase 2a VEGA (36), plus complexe dans sa méthodologie, a inclus 214 patients présentant une RCH modérée à sévère, en échec de traitements conventionnels et naïfs de traitement par anti-TNF, anti IL-12/23 et anti-IL-23. Dans cette étude, un traitement préalable par védolizumab ou tofacitinib était toléré. Les patients inclus étaient randomisés en 3 bras équilibrés, avec cette fois-ci, une combinaison (combiothérapie) de guselkumab (200 mg IV à S0, S4, S8 puis 100 mg SC/8S jusqu’à S32) et de golimumab SC (200 mg à S0, puis 100 mg à S2, S6, S10), un bras guselkumab monothérapie (IV 200 mg aux semaines S0, S4 et S8, puis 100 mg en SC toutes les huit semaines jusqu’à S32) et un bras de traitement par golimumab en injections SC à la posologie de 200 mg à S0, puis 100 mg en SC à S2, puis toutes les quatre semaines jusqu’à S34. L’objectif principal était la réponse clinique à S12. À l’inclusion, il y avait plus de malades avec une atteinte endoscopique sévère dans le bras combiothérapie (61 %), et guselkumab en monothérapie (66 %). À S12, les résultats pour la réponse clinique étaient les suivants : combiothérapie 83 %, guselkumab monothérapie 75 %, golimumab monothérapie 61 %. La différence était significative entre le bras combiothérapie et le bras golimumab monothérapie (p= 0,003 %). Pour ce qui est de l’obtention de la rémission clinique, le groupe combiothérapie était supérieur comparé aux deux autres groupes monothérapie, avec des résultats respectivement à 37 %, 21 % (p= 0,04) et 22 % (p= 0,05). À S38, il existait une efficacité supérieure pour le groupe combiothérapie versus les deux autres groupes. La rémission clinique à S38 était également obtenue dans un pourcentage significativement plus important dans le groupe combiothérapie, comparativement au groupe golimumab monothérapie (44 % versus 22 % ; p= 0,006). De la même façon les objectifs secondaires étaient plus souvent atteints à S12 et S38, dans le groupe combiothérapie pour ce qui concerne l’amélioration du score endoscopique, de la rémission histologique et du score composite de rémission histologique.
Louis et al. (37) ont récemment publié les résultats de deux études de phase 3 randomisées (2:1) versus placebo, en double aveugle, en induction (INSPIRE) et en maintenance (COMAND) du risankizumab pour le traitement de la RCH modérée à sévère. Les résultats à S12 (induction) et à S52 (maintenance) concernaient la rémission clinique définie sur la base de la fréquence des selles, des saignements, et de la cicatrisation endoscopique. Le risankizumab était prescrit à l’inclusion en IV à 1200 mg versus placebo aux semaines S0, S4 et S8. La rémission clinique à S12 était de 20,3 % (132/650) dans le groupe risankizumab comparée à 6,2 % (20/325) dans le groupe placebo (p< 0,001). Les résultats en maintenance concernaient les patients initialement traités en induction et en réponse à S12 (N= 548), puis de nouveau randomisés (1:1:1) avec une dose SC de risankizumab de 180 mg ou 360 mg toutes les 8 semaines pendant 52 semaines, en comparaison avec un groupe placebo. Le taux de rémission clinique à S52 après 12 semaines d’induction par risankizumab, était significativement supérieur dans les deux groupes traités en maintenance par risankizumab 180 mg et 360 mg versus placebo avec une efficacité respective de 40,2 % et 37,6 % et 25,1 %. Les objectifs secondaires dans les deux groupes, induction et maintenance, étaient également tous atteints pour ce qui concerne la réponse clinique, la rémission endoscopique, l’amélioration des PRO, comme les urgences intestinales ou la fatigue. Il n’y avait pas de signal particulier ou nouveau concernant les effets secondaires dans les groupes traités par risankizumab en comparaison du groupe placebo. Les effets secondaires signalés concernaient en particulier des réactions cutanées aux points d’injection, des épisodes de nasopharyngite et d’élévation modérée des enzymes hépatiques. Au vu de ces résultats, la Food and Drug Administration (FDA) américaine a approuvé le risankizumab pour le traitement de fond des formes modérées à sévères de RCH. Il est cependant à signaler que dans cette étude, les patients précédemment traités par ustékinumab ont été exclus.
Les IL-12 et 23 participent à la défense de la barrière muqueuse intestinale. Dans des modèles animaux, le déficit en IL-12 augmente le risque infectieux. Néanmoins ce surrisque infectieux n’a pas été observé chez les malades traités par ustekinumab. Les données de surveillance au long cours n’ont pas mis non plus en évidence de signal à ce sujet. Cela semble également corroboré d’un point de vue expérimental avec des souris dépourvues d’IL 23 qui restent immunocompétentes contrairement à celles totalement neutralisées pour l’IL-12 (38).
L’inhibition des interleukines paraît complexe avec des effets parfois différents voire contraires. Les anti-IL-17 semblent aggraver les MICI alors que l’inhibition de la fonction IL-23 est au contraire bénéfique et sûre (Fig. 2). Cela semble s’expliquer par le fait que même si l’IL-23 module la production d’IL-17 via les cellules de la voie Th 17, il persiste une production indépendante d’IL-17 suffisante pour garantir l’intégrité de la barrière muqueuse intestinale vis-à-vis des infections (38). S’agissant de molécules récentes, les données à long terme restent encore insuffisantes, mais celles issues des essais de phases 2 et 3, ainsi que la pharmacovigilance, sont pour le moment très rassurantes, notamment sur le plan infectieux. Nous disposons également des données en dermatologie et rhumatologie qui vont dans le même sens. Une méta analyse concernant le risankizumab pour le traitement du psoriasis chez près de 8 000 patients/années n’a mis en évidence aucun surrisque significatif sur le plan, infectieux, des cancers cutanés non mélaniques, et des risques cardiovasculaires. Pour le guselkumab, les données, disponibles à partir de données de suivi sur 5 ans vont dans le même sens (39, 40, 41).
Les données de sécurité pour les MICI sont moins nombreuses et plus récentes. Elles émanent principalement des données des études de phases 2 et 3. Les études de vraie vie sont en cours ou vont débuter (28). Les essais actuellement disponibles n’ont pas mis en évidence de signal particulier ou de surrisque significatif versus placebo, pour le traitement de la MC ou de la RCH avec le risankizumab, le mirikizumab et le guselkumab. Les données disponibles à long terme pour le risankizumab dans une étude phase 2 étendue avec une médiane de traitement de 33 mois, indique un taux d’effets secondaires sévères (principalement digestifs) de 24,6 évènements/100 patients-années avec des risques infectieux variant de 1,8 à 6,6 patients-années en fonction de leur nature. Il n’y avait aucun décès en lien avec la molécule, ni de signal d’ordre cardiovasculaire, d’infection tuberculeuse, de néoplasie ou d’infection herpétique (42).
Dans les études de phase 3 d’induction pour le traitement de la MC avec le risankizumab (ADVANCE & MOTIVATE) (26), 3 décès ont été rapportés : un décès sous risankizumab à la dose de 1200 mg (MOTIVATE) sans rapport avec la molécule, et deux décès dans les groupes placebo (ADVANCE). Trois infections sévères ont été rapportées dans l’étude MOTIVATE : une infection à E. Coli, une nasopharyngite et un sepsis. Les cinq infections rapportées dans l’étude ADVANCE n’étaient pas liées au risankizumab. Un seul patient a réactivé une tuberculose active après 8 semaines de traitement. L’étude de maintenance FORTIFY (27) n’a pas montré de surrisque d’effets secondaires versus placebo au cours des 52 semaines de suivi. Les cas incidents d’infections étaient mêmes plus faibles que dans le groupe placebo (51,4-57,7/100 patients années vs 76). Il n’y avait pas non plus de différence pour les infections sévères, toutes d’évolution favorable et sans interruption du traitement.
Dans l’essai d’induction récent INSPIRE (37) pour le traitement des formes modérées à sévères de la RCH, les effets indésirables du risankizumab les plus fréquemment signalés étaient, une infection COVID-19 (4,8 %) et une anémie (3,4 %) dans le groupe risankizumab, versus une colite ulcéreuse (10,2 %) et une anémie (6,5%) dans le groupe placebo. Le taux d’effets indésirables graves était de 2,3% pour le risankizumab par rapport à 10,2% pour le placebo. Un seul décès est survenu dans le groupe risankizumab ; il était dû à une insuffisance respiratoire causée par une pneumonie liée à l’infection COVID-19.
Dans l’essai de maintenance COMAND (37), les effets secondaires les plus fréquemment signalés parmi tous les groupes de traitement étaient, la colite ulcéreuse (13,0 % dans le groupe 180 mg de risankizumab et 13,8 % dans le groupe 360 mg de risankizumab vs 14,8 % dans le groupe le groupe placebo) et une infection COVID-19 (8,8 % dans le groupe 180 mg et 13,3 % dans le groupe 360 mg vs 11,7 % pour le placebo). La colite ulcéreuse fait référence à l’aggravation de la maladie sous-jacente, intensité de la maladie définie à la discrétion de l’investigateur.
Des événements indésirables graves ont été signalés chez 5,2 % des patients traités à 180 mg et 5,1 % dans le groupe 360 mg vs 8,2 % dans le groupe placebo. Un décès non lié au traitement est intervenu dans le groupe 360 mg, en lien avec un adénocarcinome du colon, qui préexistait à l’administration de la première dose de risankizumab. Des tumeurs malignes ont été rapportées chez 2 patients sous traitement par risankizumab (un cancer du sein canalaire invasif chez une patiente ayant des antécédents de masse mammaire et un adénocarcinome du colon avec décès non apparu sous traitement). Les deux événements n’étaient pas liés au médicament.
Avec le développement et la mise à disposition de nombreuses thérapeutiques développées au cours de ces 20 dernières années, le choix de la bonne molécule pour un patient donné s’avère de plus en plus complexe, même si les AMM nous imposent le respect de choix guidés. Sur le plan scientifique, la plupart des études ont été réalisées dans des essais comparatifs versus placebo avec peu d’essais « face-face » permettant d’évaluer directement le rapport efficacité/tolérance d’une molécule active par rapport à une autre. Certaines méta-analyses, avec des biais méthodologiques connus, ont essayé de classer l’efficacité des traitements les uns par rapport aux autres (3,43). Les méthodologies des essais récents s’avèrent par ailleurs de plus en plus complexes, hétérogènes et parfois difficiles d’analyse. En effet, les études à disposition présentent d’importantes différences méthodologiques avec :
Actuellement au vu des résultats des essais publiés, le risankizumab est le seul anti-IL-23 qui dispose d’une AMM dans le traitement des formes modérées à sévères de la maladie de Crohn, avec un périmètre de remboursement en attente. Au moins deux autres molécules sont en développement pour la même indication (mirikizumab ; guselkumab) dans des phases 2 et 3.
Le mirikizumab est le premier anti-IL-23 à avoir obtenu l’AMM dans le traitement des formes modérées à sévères de RCH, avec un périmètre de remboursement également à préciser, probablement en 3ème ligne de thérapies avancées. (Fig. 7-9)
A l’avenir, l’une des difficultés sera de bien positionner, pour un malade donné, ces nouveaux médicaments dans les différentes séquences thérapeutiques possibles en tenant compte des molécules déjà prescrites, comme les anti-TNF, l’ustekinumab, le vedolizumab et/ou les anti-JAK. Ce choix dépendra de multiples facteurs, en particulier des possibilités de remboursement (avec l’arrivée des biosimilaires), des caractéristiques de chaque patient, du rapport bénéfices/risques de chaque molécule, mais aussi d’une meilleure connaissance des mécanismes d’action et des interactions des lignes thérapeutiques les unes par rapport aux autres dans leur ordonnancement au cours du temps (4). La voie d’administration pourra également jouer un rôle dans la décision avec notamment le développement de la voie SC aussi bien en maintenance comme en induction (essai GRAVITI-Fig. 10).
A partir des données de phases 2 et 3, les anti-IL-23 sont apparus efficaces et sûrs pour le traitement de la MC. Seules des études de comparaison directe « face-face » avec une méthodologie rigoureuse permettent parfois de dégager des tendances.
En dermatologie, les anti-IL-23p19 sont apparus plus efficaces que l’adalimumab. Le guselkumab est significativement plus efficace que l’adalimumab dans une étude de phase 3 et deux essais randomisés pour le traitement du psoriasis. Dans un essai « face/face » (IMMvent), le risankizumab s’est révélé plus efficace que l’adalimumab pour le traitement du psoriasis en plaques à partir d’une évaluation du PASI (Psoriasis Area and Severity Index) (72% vs 47% à S16) (44, 45,46).
Pour ce qui concerne les MICI, les données sont plus rares. Dans l’étude VEGA pour le traitement des formes modérées à sévères de RCH, la réponse clinique et la cicatrisation endoscopique étaient comparables dans les groupes guselkumab et golimumab (36).
Dans l’étude SEAVUE comparant l’ustekinumab à l’adalimumab dans le traitement des formes naïves de biologiques et modérées à sévères de MC, aucune différence significative d’efficacité n’a été démontrée entre les deux molécules. Même s’il existait un surrisque d’infections dans le groupe adalimumab (41% vs 34%), le taux d’infections sévères était identique dans les deux groupes traités (2% vs 3%) (47).
Dans des modèles animaux, des travaux préalables ont mis en évidence que l’IL-23 contrairement à l’IL-12, était un élément essentiel du processus inflammatoire pour l’apparition spontanée de colites inflammatoires (7).
Fig. 7 : Lignes thérapeutiques possibles des anti-IL-23 selon l’AMM (Octobre 2024)
| Anti-IL-23 : AMM actuelles. Modalités d’utilisation | ||
Risankizumab* 2e ligne MC : Patients > 18 ans, avec une réponse insuffisante, ou en perte de réponse, ou intolérants aux traitements conventionnels et après le recours à au moins un anti-TNF. | Mirikizumab* 3e ligne RCH : Patients > 18 ans, avec une réponse insuffisante, ou en perte de réponse, ou intolérants aux traitements conventionnels et après le recours à au moins un anti-TNF et au vedolizumab. | |
| Induction | 600 mg perfusion IV, minimum une heure, à S0, S4 et S8. | 300 mg en perfusion IV, sur une durée ≥ 30’, à S0, S4 et S8. |
| Maintenance | 360 mg/8S à partir de S12. Injection SC avec un dispositif corporel d’injection. | 200 mg en injection SC/4S à partir de S12. Injections par 2 stylos préremplis à 100 mg chacun. |
| Réinduction | En cas de réponse insuffisante sous traitement SC : de nouveau 3 perfusions possibles de 300 mg IV à 4 semaines d’intervalle. | |
* En attente de remboursement.
| MALADIE DE CROHN : périmètre de remboursement en France en 2024 | ||
| Molécules : Biothérapies/ Nouvelles thérapies avancées (NTA) | 1re ligne de biothérapie/ NTA | 2e ligne biothérapie/ NTA |
| Infliximab | X | X |
| Adalimumab | X | X |
| Vedolizumab | X | |
| Ustekinumab | X | |
| Risankizumab* | X | |
| Upadacitinib | X | |
* Remboursement en attente
| RCH : périmètre de remboursement en France 2024 | |||
| Molécules : Biothérapies/Nouvelles thérapies avancées (NTA) | 1re ligne de biothérapie/ NTA | 2e ligne de biothérapie/ NTA | 3e ligne de biothérapie/ NTA |
| Infliximab Adalimumab Golimumab | X X X | X X X | X X X |
| Vedolizumab | X | X | X |
| Ustekinumab | X | X | |
| Tofacitinib Filgotinib Upadacitinib | X X X | ||
| Mirikizumab* | X | ||
* Remboursement en attente
Désormais, il apparaît nécessaire de concevoir des études nouvelles comparatives en face – face, afin de démontrer que la neutralisation sélective de l’IL-23 est plus efficace que le blocage combiné de l’IL-12 et de l’IL-23 dans les pathologies inflammatoires intestinales. Dans le traitement du psoriasis une étude de phase 3 en face – face, comparant le risankizumab versus l’ustékinumab pour le traitement des formes modérées à sévères du psoriasis en plaque, a clairement démontré une supériorité du risankizumab (18).
A partir de ces résultats en dermatologie il est apparu initialement difficile de conclure à une supériorité du guselkumab versus l’ustékinumab pour le traitement de la maladie de Crohn, notamment à partir des résultats de l’étude GALAXI 1 de phase 2, en raison d’un trop faible effectif de patients inclus dans le bras ustékinumab. Ainsi, il n’y avait pas de différence statistiquement significative entre les deux traitements dans l’évaluation globale des patients à S12, aussi bien sur la rémission clinique (53 % versus 46 %), les PRO2 (42,7 % versus 39,7 %), la réponse endoscopique (35,7 % versus 28,6 %) et les marqueurs de réponse bioclinique (47 % versus 46 %). De la même façon l’évaluation clinique sur les PRO2 à S48 ne permettait pas de mettre en évidence de différence significative entre le guselkumab et l’ustékinumab (24,25).
De façon plus récente, l’étude SEQUENCE (48) en face/face, de phase 3b, en simple aveugle, multicentrique et randomisée, a comparé directement l’efficacité et la tolérance du risankizumab et de l’ustékinumab chez 520 patients atteints d’une MC modérée à sévère. Les malades inclus étaient tous en échec d’au moins un traitement par anti-TNF et randomisés avec un ratio 1:1 entre les groupes risankizumab ou ustekinumab. Les critères de jugement principaux étaient la rémission clinique à S24 (non infériorité du risankizumab par rapport à l’ustékinumab) et la rémission endoscopique à S48 (supériorité du risankizumab). La rémission clinique à S24 était obtenue chez 58,6% des patients pour le risankizumab vs 39,5% pour l’ustekinumab. La rémission endoscopique à S48 était obtenue chez 31,8 % pour le risankizumab vs 16,2 % pour l’ustekinumab. Tous les critères secondaires étaient également atteints. L’incidence globale des effets indésirables était identique dans les deux groupes. Les évènements indésirables graves ou responsables d’un arrêt de l’étude, étaient inférieurs avec le risankizumab en comparaison de l’ustekinumab. D’un point de vue méthodologique, même si cette étude était en ouvert, la relecture endoscopique centralisée a été réalisée en aveugle et tous les critères évalués étaient par ailleurs clairement en faveur du risankizumab.
Au vu des résultats de cette étude, la HAS a rendu un avis favorable en mars 2024 (29) pour le remboursement du risankizumab en 2ème ligne de biothérapie, pour le traitement de la MC active, modérée à sévère, chez les patients adultes en échec (réponse insuffisante, perte de réponse ou intolérance) d’un traitement conventionnel par corticoïdes, immunosuppresseurs et d’au moins un anti-TNF ou de contre-indication à l’un ces traitements. Son remboursement est en attente en France.
Fig. 10 : Principales études d’évaluation des anti-IL-23.
| ETUDES publiées et en cours/ Anti-IL-23 | Phase 2 | Phase 3 | Randomisée | Comparateur actif | Vraie vie |
| Maladie de Crohn | |||||
| SERENITY (N=191) Mirikizumab | X | X | |||
| VIVID 1 (N =1150) Mirikizumab. V. Jairath et al. | X | X | X (vs placebo/ ustekinumab) | ||
| GALAXI 1 (N = 309) Guselkumab: Induction. WJ. Sandborn et al. Maintenance. S.Danese et al. | X | X | X (vs placebo/ ustekinumab) | ||
| GALAXI 2 & 3 (N = 508 ; N = 513) Guselkumab. R. Panacionne et al. | X | X | X (vs placebo/ ustekinumab) | ||
| GRAVITI Guselkumab induction SC R. Panacionne et al. | X | X | |||
| FUSION CD. Guselkumab | X | X | |||
| Risankisumab (N = 121). Feagan et al. | X | X | |||
| ADVANCE (N = 191). Risankizumab induction. D’Haens et al. | X | X | |||
| MOTIVATE (N = 618). Risankizumab induction. D’Haens et al. | X | X | |||
| FORTIFY (N = 542). Risankizumab maintenance | X | X | |||
| Risankizumab (N = 174). Fumery et al. | X (rétrospective) | ||||
| SEQUENCE (N = 520) Risankizumab. Peyrin Biroulet et al. | X | X | X (vs ustekinumab) |
| RCH | |||||
| INSPIRE (N = 977) Risankizumab induction. Louis et al. | X | X | |||
| COMMAND (N = 584) Risankizumab maintenance. Louis et al. | X | X | |||
| LUCENT 1 (N = 1281) Mirikizumab(N =) induction. G. D’Haens et al. | X | X | |||
| Lucent 2 (N = 544) Mrikizumab maintenance. G. D’Haens et al. | X | X | |||
| QUASAR (N = 313) Guselkumab induction. Peyrin-Biroulet et al. | X 2b | X | |||
| QUASAR (N = 701). Guselkumab maintenance. D. Rubin et al. | X | X | |||
| VEGA (N = 214) Guselkumab combiothérapie-Golimumab vs Guelkumab vs placebo Feagan et al. | X Preuve de concept | X |
Les résultats de cette étude importante suggèrent une supériorité potentielle intrinsèque des anti-IL-23 par rapport aux anti-IL12/23 dans le traitement des MC modérées à sévères. En faveur de cette potentielle supériorité d’efficacité, en dehors de voies de signalisation immunes privilégiées, d’autres différences peuvent être évoquées, comme une biodisponibilité supérieure au niveau de la muqueuse intestinale inflammatoire et une meilleure affinité au niveau des cibles cellulaires responsables d’interrégulations entre les différentes cellules immunitaires. Par ailleurs, il a été également montré que l’efficacité des anti-IL-23 était sans doute majorée au niveau de certaines cellules immunitaires devenues résistantes aux anti-TNF (4), ce qui était l’une des caractéristiques des patients inclus dans l’étude SEQUENCE.
En conséquence, d’autres essais de confirmation seront nécessaires pour mieux déterminer les sous-groupes de patients pouvant justifier d’une indication privilégiée d’anti-IL-23 par rapport aux autres biothérapies actuellement disponibles.
À partir des données des études de phase 2 et 3 concernant le risankizumab, le mirikizumab et le guselkumab (Fig. 10), il existe des arguments en faveur de l’efficacité et de la sécurité de ces molécules pour maintenir la rémission clinique et endoscopique chez les patients naïfs ou non de biothérapies présentant une MC modérée à sévère (48).
À partir des analyses en sous-groupes des études ADVANCE, MOTIVATE et FORTIFY (26,27), il est apparu que l’efficacité du risankizumab était maintenue indépendamment des échecs des lignes de biothérapies précédentes.
Dans une méta-analyse en réseau récente (49), les données cumulées et analysées à partir de 25 études concernant plus de 8700 patients atteints de MC modérée à sévère, l’infliximab 5mg/kg, restait la molécule classée en premier pour l’induction et la rémission clinique en comparaison des autres classes thérapeutiques de biothérapies. Néanmoins, pour ce qui est de l’analyse des résultats des patients déjà bio exposés, le risankizumab à la posologie de 600 mg était la molécule la plus efficace pour l’induction et la rémission clinique lorsque les malades bio naïfs et bio exposés étaient regroupés dans l’analyse. Bien entendu, les résultats des méta-analyses doivent être interprétés avec précaution en raison de multiples biais potentiels concernant le caractère hétérogène des populations étudiées et des critères d’inclusion. Néanmoins, les critères d’inclusion concernant les nouvelles molécules ont permis de sélectionner en général des populations de patients plus sévères et en échec de lignes précédentes de biothérapies. Dans les études de phase 3 concernant l’évaluation de l’efficacité du risankizumab, plus de 40 % de patients étaient en échec de deux ou plus de biothérapies.
En conséquence, et à l’aune de ces derniers résultats, les anti-IL-23 pourraient être positionnés dès la 2ème ligne de biothérapie dans le traitement de la maladie de Crohn (Fig. 7, 8).
Les études de phase 3 concernant les données d’efficacité et de tolérance des anti- IL-23 dans le traitement de la RCH sont encore limitées. Les premières données orientent vers une efficacité et une très bonne tolérance chez les patients bio naïfs et bio exposés. Dans l’étude VEGA (36), il n’apparaissait pas de différence clairement significative entre le guselkumab et le golimumab chez les patients naïfs d’anti-TNF pour le traitement de la RCH modérée à sévère. Actuellement il pourrait être envisagé de proposer cette classe thérapeutique au minimum en 3ème ligne de biothérapies ou de thérapies avancées (Fig. 7, 9).
À partir des données expérimentales, des méta-analyses, des études de phases 2/3 récentes et des premières études de vraie vie (Fig. 10), il apparaît que les anti-IL-23, avec actuellement trois molécules (risankizumab, mirikizumab et guselkumab), sont efficaces dans le traitement des formes modérées à sévères de MC et de RCH, aussi bien chez les patients bio naïfs que bio exposés. Moins immunogènes et plus persistantes, leur efficacité apparaît proche de celle des anti-TNF et supérieure en cas d’échec thérapeutique de lignes précédentes de biothérapies. La tolérance paraît excellente et l’immunogénicité faible. Les premières données semblent montrer à partir d’études de comparaisons directes, une efficacité supérieure des anti-IL-23 versus les anti-IL-12/23, ce qui pourrait faire positionner cette classe thérapeutique dès la 2e ligne des NTA, au moins pour la MC. De nouvelles études de comparaisons et de positionnements de lignes thérapeutiques seront à concevoir également avec les autres NTA actuelles comme les anti-JAK et les modulateurs de S1P. Ces trois anti-IL-23 disposent déjà, ou disposeront d’une AMM européenne pour le traitement des MICI. L’obtention du remboursement en France et la détermination de leur positionnement par rapport aux autres drogues disponibles, seront alors discutées à l’occasion d’une négociation du prix auprès des pouvoirs publics à l’aune des résultats des dernières études. Dans un avenir plus ou moins proche, au vu de leur profil d’efficacité et de tolérance favorable, des évolutions dans leurs stratégies d’utilisation sont encore à attendre, comme l’induction en SC, l’administration per os avec une galénique adaptée, et de nouvelles associations avec d’autres biothérapies ou thérapies avancées.
FMC HGE : Organisme certifié Qualiopi pour la catégorie ACTIONS DE FORMATION.
