LIENS D’INTÉRÊT
TAKEDA : consultant, orateur – CSL Behring : consultant – GRIFOLS : orateur
MOTS-CLÉS
Cholestase néonatale ; fibrose hépatique ; cirrhose ; transplantation hépatique
ABRÉVIATIONS
AAT : alpha1-antitrypsine ;
AFP : alpha-foetoproétine ;
ALAT : alanine aminotransférase ;
APRI : AST-to-platelet ratio index ;
AUDC : acide ursodésoxycholique ;
BPCO : broncho-pneumopathie chronique obstructive ;
CAP : controlled attenuation parameter ;
CHC : carcinome hépatocellulaire ;
DAAT : déficit en alpha1-antitrypsine ;
DLCO : capacité de diffusion du monoxyde de carbone ;
EASL : European Association for the Study of Liver disease ;
Fib-4 : Fibrosis-4 index ;
GGT : gamma-glutamyl transférase ;
HTP : hypertension portale ;
IEF : iso-électro focalisation ;
INR : International Normalized Ratio ;
PAS-D+ : coloration par l’acide périodique de Schiff et persistant après digestion par la diastase ;
PCR : polymerase chain reaction ;
SPLF : Société de Pneumologie de Langue Française ;
TH : transplantation hépatique ;
VEMS : volume expiratoire maximal par seconde ;
TP : taux de prothrombine.
Introduction
Le déficit en alpha1-antitrypsine (DAAT) est une maladie génétique concernant tous les âges et prédisposant à une atteinte hépatique et pulmonaire, de sévérité variable (1). C’est une maladie rare, mais encore largement sous-diagnostiquée, alors qu’une prise en charge adaptée permet d’en améliorer le pronostic. Elle est liée à la présence de variants pathogènes du gène SERPINA1 codant pour l’alpha1-antitrypsine (AAT). Le variant Z à l’état homozygote est responsable du génotype ZZ, prédisposant à la forme la plus sévère de la maladie, avec des manifestations hépatiques dès la période néonatale et un risque de fibrose augmentée à l’âge adulte, dont le seul traitement curatif est la transplantation hépatique (TH). L’atteinte pulmonaire, aussi variable et responsable d’une altération de la qualité de vie, est accessible à un traitement substitutif (1-3). La présence du variant Z à l’état hétérozygote augmente le risque de fibrose hépatique, notamment en cas d’association à d’autres cofacteurs de fibrose hépatique (2, 4, 5). De nouvelles thérapeutiques, notamment les ARN interférents, sont en cours de développement, avec des résultats prometteurs ouvrant la voie à de nouvelles stratégies thérapeutiques (2). Des recommandations de bonnes pratiques cliniques ont été établies en 2024 par l’European Association for the Study of Liver disease (EASL) pour la prise en charge diagnostique et thérapeutique de l’atteinte hépatique liée au DAAT (6).
Physiopathologie
L’AAT est une glycoprotéine de la famille des serpines, produite principalement dans les hépatocytes en cas d’inflammation et sécrétée dans la circulation sanguine. Elle est d’une part une anti-protéase, inhibant les sérines protéases, notamment l’élastase sécrétée par les neutrophiles et protégeant ainsi les tissus de la dégradation et un puissant agent anti inflammatoire. Son principal tissu cible est le poumon (1). La présence de variants pathogènes du gène SERPINA1, codant pour l’alpha1-antitrypsine, est responsable d’un mauvais repliement de cette protéine et d’anomalies de sa dégradation au niveau du réticulum endoplasmique des hépatocytes : on parle alors de « gain de toxicité », responsable d’un stress protéotoxique générant des lésions hépatiques parfois sévères (7). La diminution de l’AAT dans la circulation sanguine entraîne une « perte de fonction », l’accumulation notamment d’élastase neutrophilique, favorisant la sécrétion de mucine et de protéines de l’inflammation, responsable de l’atteinte pulmonaire, se manifestant par de l’emphysème pulmonaire pan-lobulaire et/ou une bronchopneumopathie chronique obstructive (1).
Les variants de l’AAT sont classés selon leur vitesse de migration en iso-électrofocalisation, le variant sauvage étant noté Pi*M (vitesse moyenne = normale), les variants pathologiques les plus courants étant Pi*Z (very slow) ou Pi*S (slow), mais plus de 600 variants sont décrits, avec une expression co-dominante des 2 allèles (8). Ainsi, le génotype « normal » est MM, permettant la sécrétion normale dans la circulation sanguine d’une AAT fonctionnelle à un taux supérieur à 1,1 g/L. La forme la plus sévère du DAAT est caractérisée par la présence de l’allèle Pi*Z (Glu342Lys = substitution d’un glutamate par une lysine en position 342) à l’état homozygote (génotype ZZ), conduisant à la production d’une AAT mal repliée (AAT-Z), s’agrégeant sous forme de polymères dégradés dans l’hépatocyte par les voies de l’autophagie et du protéasome (1). Ces polymères dégradés dans le réticulum endoplasmique conduisent à des inclusions d’AAT, visibles en histologie grâce à la coloration par l’acide périodique de Schiff et persistant après digestion par la diastase (PAS-D+), ce qui est caractéristique de cette pathologie (1). Le taux sanguin d’AAT en cas de génotype ZZ est généralement inférieur à 0,57 g/L. Un autre variant relativement fréquent peut être retrouvé, Pi*S (Glu264Val), et causer un déficit moins sévère, mais tout de même significatif lorsqu’il est associé au variant Pi*Z (génotype SZ) (9).
Épidémiologie
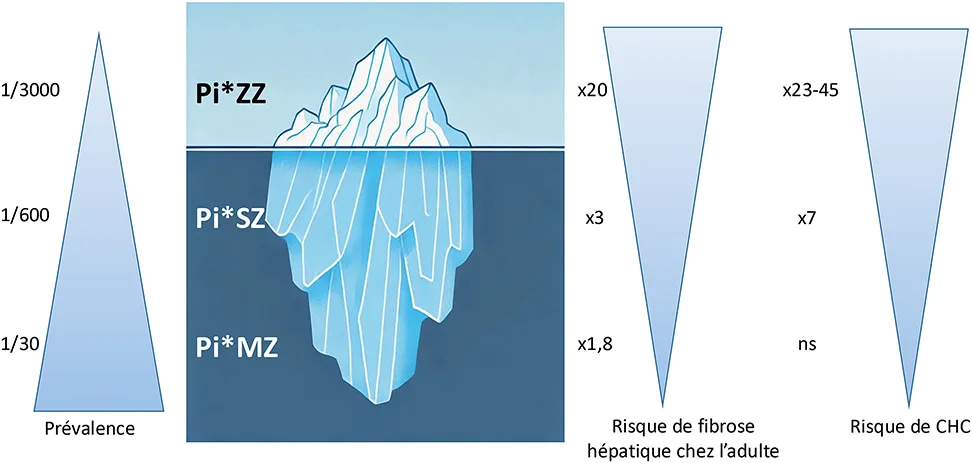
La forme classique du DAAT (génotype ZZ) est une maladie rare qui affecte environ 1 personne sur 3 000, tandis que la présence à l’état hétérozygote de l’allèle Pi*Z (génotype MZ) est retrouvée chez une personne sur 30 d’origine européenne (4 % de la population) (figure 1) (10). L’allèle Pi*S est retrouvé chez une personne sur 20, notamment chez les personnes originaires de la péninsule ibérique. Les autres allèles, et donc les différents génotypes qui en résultent sont plus rares (11).
Atteinte hépatique chez l’enfant
L’atteinte hépatique chez l’enfant est d’une grande variabilité (2). Environ 10 % des patients Pi*ZZ présentent une cholestase néonatale, caractérisée par un ictère dans les premières semaines de vie, des selles souvent décolorées et des urines foncées, confirmée par une hyperbilirubinémie conjuguée et une élévation de la gamma-glutamyl transférase (GGT), des transaminases et des acides biliaires (12, 13).
Figure 1 : Épidémiologie et atteinte hépatique des phénotypes de déficit en alpha1-antitrypsine [adapté de (4)

À ce stade, la démarche diagnostique doit systématiquement écarter les diagnostics différentiels, tels que l’atrésie des voies biliaires, le syndrome d’Alagille, la mucoviscidose, les causes infectieuses, mais aussi les autres causes de cholestases intra-hépatiques génétiques (14). Souvent, les patients sont asymptomatiques et le diagnostic est fait plus tardivement à l’occasion de perturbations du bilan hépatique, ce qui est le cas de plus de la moitié des patients, ou d’une hépatosplénomégalie. Pour la majorité des patients Pi*ZZ, l’évolution est favorable dans l’enfance, avec un bilan hépatique normal ou subnormal à la fin de l’adolescence, mais de rares patients (< 5 %) vont présenter une maladie hépatique sévère pouvant nécessiter une TH dans l’enfance (15, 16). Les facteurs de mauvais pronostic chez l’enfant sont la persistance d’une cholestase, une élévation des alanine aminotransférase (ALAT), de l’International Normalized Ratio (INR), ainsi qu’une hypertension portale (HTP) progressive (2). Les facteurs modificateurs responsables de la variabilité de l’atteinte hépatique chez l’enfant, y compris au sein de mêmes fratries, ne sont pas élucidés et impliquent probablement des polymorphismes génétiques, des différences de transport protéique, ainsi que des régulations différentes dans les mécanismes de dégradation par l’autophagie ou le protéasome (17-21).
Bien qu’elle soit rarement réalisée, la biopsie hépatique peut montrer une variété de lésions non spécifiques chez le nourrisson, comme une paucité ductulaire et une prolifération cholangiolaire, des cellules géantes, une stéatose, une hépatite lobulaire ou une fibrose portale (22). Les globules d’AAT sont visualisés chez des enfants plus âgés, et ne semblent pas corrélés à l’évolution de l’atteinte hépatique contrairement à l’adulte.
En raison de la variabilité de l’atteinte hépatique, le suivi doit être adapté selon la sévérité, avec la réalisation au moins annuellement pour les patients Pi*ZZ d’un bilan hépatique, d’une échographie et d’une mesure non-invasive de la fibrose (2). La mesure de l’élastographie hépatique est de plus en plus réalisée chez l’enfant, avec une bonne faisabilité surtout après 2 ans et permettant une bonne corrélation avec la sévérité de l’atteinte hépatique (23, 24). Les patients ayant une atteinte hépatique sévère doivent être orientés vers un centre de référence en hépatologie pédiatrique, pour prendre en charge les complications de l’HTP et déterminer le moment approprié pour envisager une TH, celle-ci étant le seul traitement curatif disponible (1, 2, 25, 26).
Chez l’enfant, le génotype hétérozygote Pi*MZ n’est généralement pas suffisant pour développer une atteinte hépatique significative (2). Il peut néanmoins se manifester par une cholestase néonatale transitoire, des perturbations légères et fluctuantes du bilan hépatique ou une hépatomégalie (12). Il s’agit également d’un cofacteur pouvant aggraver d’autres maladies hépatiques telles que l’atrésie des voies biliaires ou la mucoviscidose (27, 28). Le génotype Pi*SZ mais aussi d’autre génotypes plus rares peuvent aussi être responsables d’atteinte hépatique sévère (12, 13).
Un point clé de la prise en charge de cette hépatopathie chronique est la transition vers l’hépatologie adulte (2). Celle-ci se répand largement dans la prise en charge des maladies chroniques à début pédiatrique, et ses modalités sont de plus en plus codifiées (29). Elle dépend de plusieurs éléments tels que la sévérité de l’atteinte hépatique, la maturité de l’adolescent, ainsi que de l’environnement social, familial, scolaire et professionnel. Elle est assez évidente en cas d’atteinte sévère ou d’antécédent de TH, mais demeure plus compliquée lorsque le patient est asymptomatique, d’autant que l’histoire naturelle du DAAT est peu connue entre l’adolescence et la 4e ou 5e décennies. Cette transition doit se préparer dès l’adolescence, notamment avec les conseils hygiéno-diététiques de prévention (alcool, tabac, alimentation), et elle doit s’articuler conjointement entre le pédiatre et l’hépatologue adulte (30). Dans le DAAT, une première consultation auprès d’un pneumologue d’adulte est conseillée à ce moment-là avec une exploration de la fonction respiratoire et des conseils de prévention du tabagisme.
Atteinte hépatique chez l’adulte
La majorité des adultes déficitaires en AAT ont un bilan hépatique normal, mais certains peuvent avoir une élévation des transaminases et GGT, notamment en cas de génotype Pi*ZZ (4, 31). Un bilan étiologique différentiel doit être réalisé pour rechercher d’autres comorbidités (2). Même en cas de perturbations minimes de la biologie hépatique, les patients DAAT ont un risque augmenté de développer une fibrose hépatique avancée, 20 fois supérieur en cas de génotype Pi*ZZ, 3 fois supérieur pour le Pi*SZ et près de 2 fois pour les Pi*MZ (4, 31). C’est pourquoi il est recommandé de réaliser une mesure non invasive de la fibrose au diagnostic, par élastographie ou en utilisant les scores biologiques de fibrose tel que l’APRI (AST-to-platelet ratio index) ou le Fib-4 (Fibrosis-4 index) (24, 32). Une valeur d’élasticité hépatique mesurée par FibroScan® au-delà de 7,1 kPa permet de détecter une fibrose significative (32-35). Une stéatose hépatique peut également être présente chez certains patients, et peut être évaluée par le CAP (controlled attenuation parameter), son caractère prédictif de fibrose hépatique en l’absence d’autre cofacteur n’est pas confirmé (32, 33).
Une biopsie hépatique est rarement indiquée, elle doit être considérée en cas de difficulté d’évaluation non-invasive de la fibrose ou lorsqu’un diagnostic différentiel doit être éliminé (2, 6).
Comme chez l’enfant, l’atteinte hépatique du génotype Pi*ZZ est variable : entre 20 et 36 % des patients vont développer une fibrose hépatique significative, et plus de 10 % une cirrhose même en l’absence de comorbidités (4, 33, 35). Les facteurs de risque identifiés sont le sexe masculin, l’âge supérieur à 50 ans et la présence d’un syndrome métabolique (35). Une autre caractéristique importante est le risque de carcinome hépatocellulaire (CHC) 20 à 50 fois supérieur chez les patients Pi*ZZ, notamment en cas de fibrose avancée, qui justifie donc son dépistage tous les 6 mois (2, 31, 36).
Le suivi est donc proposé selon la sévérité de l’atteinte hépatique et la présence de comorbidités, tous les 1 à 3 ans en cas d’atteinte légère à modérée, et au moins semestriel en cas de fibrose avancée, sans oublier le dépistage du CHC par échographie et dosage de l’alpha-fœtoprotéine (AFP) (2).
L’allèle Pi*Z est considéré comme un gène modificateur, y compris à l’état hétérozygote, avec un risque augmenté d’événements hépatiques, tels qu’une décompensation plus rapide d’hépatopathie chronique ou un décès de cause hépatique, un risque augmenté de fibrose hépatique en cas de maladie hépatique stéatosique alcoolique ou non-alcoolique (2, 37). Aussi, la présence de cofacteurs métaboliques tels qu’une obésité, un diabète ou un syndrome métabolique chez les sujets Pi*MZ augmente le risque d’avoir une fibrose hépatique significative (5). Cette augmentation du risque de fibrose a été décrite dans d’autres pathologies hépatiques (hépatites B, C, hémochromatose), sur de petits effectifs, et doit être confirmée à plus large échelle (4). Enfin, un sur-risque de présenter une maladie lithiasique hépatique est rapporté chez les sujets Pi*MZ (OR 1,3) (38).
En raison de ce risque augmenté d’atteinte hépatique chez les sujets hétérozygotes, et en l’absence de données évaluant systématiquement le devenir de patients greffés avec un foie de donneur hétérozygote Pi*MZ, les greffons de donneurs Pi*MZ peuvent être considérés pour une TH qu’en l’absence d’autre possibilité et de lésions hépatiques sur le greffon (6). Les greffons hépatiques de donneurs homozygotes Pi*ZZ, par contre, ne doivent pas être utilisés en routine.
Évaluation de l’atteinte pulmonaire
L’atteinte pulmonaire liée au DAAT est caractérisée par un emphysème pan-lobulaire prédominant habituellement aux bases, responsable d’une broncho-pneumopathie chronique obstructive (BPCO), avec toux, dyspnée et exacerbations, et aggravée par le tabagisme (1, 3). Elle va concerner les patients de plus de 50-60 ans, et plus tôt en cas de tabagisme. Cette atteinte est variable et non spécifique, expliquant l’absence de diagnostic de cette pathologie dans environ 90 % des cas, avec un retard diagnostique de 5 à 7 ans après le début des symptômes (39). Le scanner thoracique permet de mettre en évidence l’emphysème, et les explorations fonctionnelles respiratoires révèlent une altération de la mesure du volume expiratoire maximal par seconde (VEMS) et de la capacité de diffusion du monoxyde de carbone (DLCO). Un diagnostic précoce permet de délivrer des conseils de prévention, d’hygiène de vie ainsi qu’un traitement adapté, afin d’améliorer la qualité de vie (40).
Diagnostic
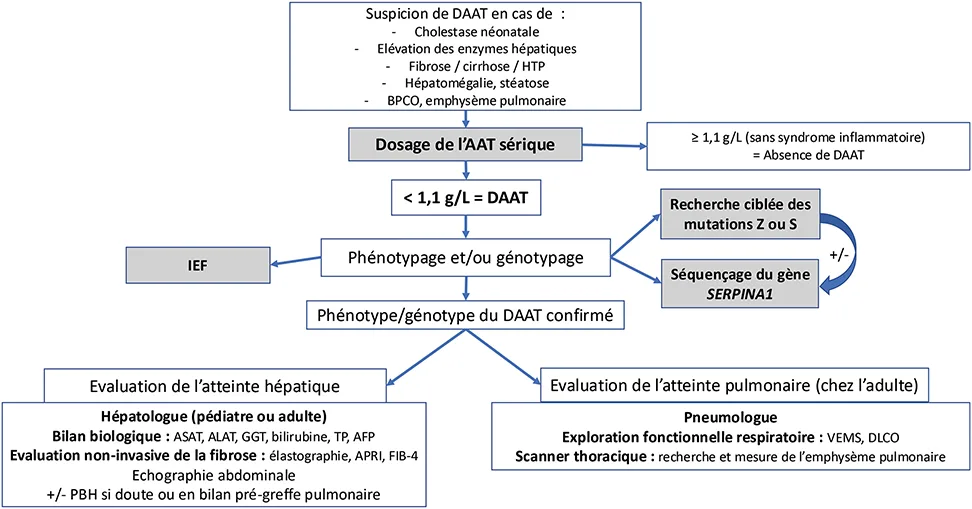
Un DAAT doit être évoqué dans différentes circonstances, mais cette pathologie est encore largement sous-diagnostiquée (figure 2) (2, 24, 39). Chez l’enfant, il doit être recherché en cas de cholestase néonatale ou de perturbation chronique du bilan hépatique, ainsi qu’en cas de fibrose hépatique comme chez l’adulte, a fortiori en cas de cirrhose. En raison du risque plus élevé de décompensation hépatique, y compris en cas de phénotype hétérozygote, la recherche d’un DAAT doit également être réalisée chez tout patient présentant une hépatopathie chronique. Ce diagnostic doit être évoqué chez tout patient adulte présentant une bronchopneumopathie chronique, et notamment en cas d’emphysème pulmonaire, peu importe l’âge ou le statut tabagique du patient, ainsi qu’en cas d’asthme réfractaire aux traitements (41). L’atteinte pulmonaire ne se manifeste pas avant la 3e ou 4e décennie, il n’y a donc pas d’indication à rechercher ce diagnostic chez un enfant présentant une symptomatologie respiratoire. Un dépistage familial peut également être proposé, notamment pour la fratrie d’un patient homozygote, ou en cas de manifestations hépatiques ou pulmonaires pour les apparentés (parents ou enfants d’un cas index). D’autre situations plus rares doivent faire évoquer un DAAT, telle qu’une panniculite ou une vascularite à c-ANCA (1). Le diagnostic est parfois fortuit à l’occasion d’une électrophorèse des protéines mettant en évidence une diminution du pic d’alpha1-globulines, ou à l’occasion d’un test génétique plus large tel qu’un exome ou un génome.
Figure 2 : Diagnostic du déficit en alpha1-antitrypsine [adapté de (24)

Lorsque le diagnostic est évoqué, la première étape est de doser la concentration sérique d’AAT (6, 41). Une concentration < 1,1 g/L est considérée comme basse, notamment en l’absence de syndrome inflammatoire biologique, et les phénotypes les plus sévères ont généralement une concentration < 0,50 g/L (6). Lorsque la concentration est basse ou douteuse notamment en cas de syndrome inflammatoire, le phénotype doit être précisé, soit par iso-électro focalisation (IEF), soit par génotypage, selon la disponibilité des techniques de laboratoire. L’IEF permet d’identifier les phénotypes liés aux allèles les plus fréquents (Pi*M, Pi*Z, Pi*S) mais requiert une expertise du biologiste (3). Pour le génotypage, on peut rechercher de manière ciblée les mutations les plus fréquentes par polymerase chain reaction (PCR) (Pi*Z et Pi*S, voire plus avec des panels élargis), ou réaliser un séquençage du gène SERPINA1 en cas de discordance entre la concentration et le phénotype afin d’identifier des variants rares. Il est important de noter que le test PCR et le séquençage du gène sont des tests génétiques, donc soumis à des règles de prescriptions, et qui doivent être réalisés avec précaution, notamment en cas de dépistage familial (3).
Lorsque le diagnostic de DAAT est confirmé, un avis spécialisé est recommandé pour évaluer l’atteinte hépatique et pulmonaire, pour le suivi, pour la prise en charge, ainsi que la mise en place d’un traitement éventuel (24). En France, le DAAT fait partie des filières de santé maladies rares FILFOIE et RESPIFIL, permettant d’orienter les patients vers des centres de référence ou de compétence de cette pathologie.
Traitement de l’atteinte hépatique
Chez l’enfant, en cas de cholestase, une prise en charge nutritionnelle est indispensable avec une supplémentation en vitamines liposolubles, et un traitement par acide ursodésoxycholique (AUDC) peut être proposé mais les données sont insuffisantes à ce jour pour le recommander formellement (6, 42). Chez certains enfants, l’AUDC permet d’améliorer le bilan hépatique mais sans effet sur l’évolution des patients les plus sévères (43).
L’accumulation d’AAT-Z dans les hépatocytes est exacerbée par les autres facteurs d’atteinte hépatique tel que le surpoids et l’alcool, qui doivent donc être limités chez les patients déficitaires (6). En cas d’atteinte hépatique sévère, tout comme dans toute hépatopathie, la consommation d’alcool doit être évitée. La vaccination contre l’hépatite A et B doit également être proposée en fonction du statut sérologique, comme à tout patient atteint d’hépatopathie chronique.
Il n’y a pas de traitement approuvé pour l’atteinte hépatique du DAAT à ce jour, en dehors de la TH en cas d’hépatopathie sévère (25). Celle-ci a un bon pronostic dans cette indication, chez les enfants comme les adultes, notamment en l’absence d’atteinte pulmonaire avancée (26). La TH permet de normaliser la concentration sérique d’AAT, mais à ce jour peu de données existent sur l’évolution au long cours de la fonction respiratoire chez les patients greffés hépatiques (2, 44, 45).
Traitement de l’atteinte pulmonaire
En plus de la prise en charge classique de la BPCO tels que l’utilisation de bronchodilatateurs, la réhabilitation respiratoire, une oxygénothérapie longue durée si nécessaire, associés à un sevrage tabagique, une supplémentation intraveineuse en alpha1-antitrypsine peut être indiquée (3, 39, 46). Ce traitement dérivé du plasma permet de réduire le déclin de la densité pulmonaire, mais n’a pas montré à ce jour, d’effet sur le VEMS, la qualité de vie ou les exacerbations de BPCO. En France, sa prescription fait l’objet de recommandations établies par la Société de Pneumologie de Langue Française (SPLF), voire de discussion en réunion de concertation multidisciplinaire (47).
Perspectives thérapeutiques pour l’atteinte hépatique
L’amélioration des connaissances physiopathologiques de cette pathologie au cours des dernières décennies ouvre la voie au développement de thérapeutiques prometteuses (1, 2, 4, 48, 49). On peut ainsi agir en diminuant la production d’AAT-Z, en améliorant son repliement, en stimulant sa dégradation dans les hépatocytes ou sa sécrétion dans la circulation sanguine. L’inhibition de la production d’AAT-Z est actuellement possible par l’ARN interférent Fazirsiran permettant de diminuer la concentration sérique et intra-hépatique d’AAT-Z, associé à une amélioration de la biologie hépatique, ainsi qu’à une amélioration de la fibrose hépatique chez certains patients, avec une sécurité sur le plan pulmonaire sur la durée de l’étude (50, 51). Ce traitement est actuellement évalué chez l’adulte dans un essai clinique multicentrique de phase 3 (NCT05677971). Un autre ARN interférent, Belcesiran, est actuellement évalué dans un essai clinique de phase 2 (NCT04764448). Des approches d’édition du génome (CRISPR-Cas9) ou d’édition de base au niveau de l’ADN et de l’ARN sont en développement avec des essais cliniques en cours de mise en place (NCT06622668 ; NCT06389877 ; NCT06405633 ; NCT06186492) (52). Enfin, le repliement de la protéine peut être amélioré par des protéines chaperonnes, la polymérisation de l’AAT-Z peut être bloquée par des petits peptides ou des anticorps intracellulaires (intrabodies), et l’autophagie peut être stimulée par des drogues telles que la carbamazépine ou le sirolimus (2, 48).
Conclusion
Bien qu’étant une hépatopathie rare, le DAAT représente un modèle de pathologie hépatique monogénique, dont les perspectives de traitement augmentent avec l’amélioration des connaissances de la physiopathologie et de l’histoire naturelle. Une amélioration du diagnostic et de l’évaluation des atteintes hépatiques et pulmonaires permettrait de proposer des prises en charge thérapeutiques individualisées aux patients déficitaires. Enfin, une diminution des facteurs de risque hépatique est justifiée chez les sujets hétérozygotes en raison d’un risque accru de fibrose hépatique.
Références
- Strnad P, McElvaney NG, Lomas Alpha1-Antitrypsin Deficiency. N Engl J Med. 09 2020;382(15):1443-55.
- Ruiz M, Lacaille F, Schrader C, Pons M, Socha P, Krag A, et Pediatric and Adult Liver Disease in Alpha-1 Antitrypsin Deficiency. Semin Liver Dis. août 2023;43(3):258-66.
- Mornex JF, Traclet J, Guillaud O, Dechomet M, Lombard C, Ruiz M, et Alpha1-antitrypsin deficiency: An updated review. Presse Med. sept 2023;52(3):104170.
- Fromme M, Schneider CV, Trautwein C, Brunetti-Pierri N, Strnad Alpha-1 antitrypsin deficiency: A re-surfacing adult liver disorder. J Hepatol. avr 2022;76(4):946-58.
- Schneider CV, Hamesch K, Gross A, Mandorfer M, Moeller LS, Pereira V, et al. Liver Phenotypes of European Adults Heterozygous or Homozygous for Pi*Z Variant of AAT (Pi*MZ vs Pi*ZZ genotype) and Non-carriers. Gastroenterology. 3 mai 2020;
- European Association for the Study of the EASL Clinical Practice Guidelines on genetic cholestatic liver diseases. J Hepatol. août 2024;81(2):303-25.
- Lomas DA, Evans DL, Finch JT, Carrell The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature. 18 juin 1992;357(6379):605-7.
- Ferrarotti I, Thun GA, Zorzetto M, Ottaviani S, Imboden M, Schindler C, et Serum levels and genotype distribution of a1-antitrypsin in the general population. Thorax. août 2012;67(8):669-74.
- Mahadeva R, Chang WS, Dafforn TR, Oakley DJ, Foreman RC, Calvin J, et Heteropolymerization of S, I, and Z alpha1-antitrypsin and liver cirrhosis. J Clin Invest. avr 1999;103(7):999-1006.
- de Serres FJ, Blanco I, Fernández-Bustillo PI S and PI Z alpha-1 antitrypsin deficiency worldwide. A review of existing genetic epidemiological data. Monaldi Arch Chest Dis. déc 2007;67(4):184-208.
- Ferrarotti I, Wencker M, Chorostowska-Wynimko Rare variants in alpha 1 antitrypsin deficiency: a systematic literature review. Orphanet J Rare Dis. 22 févr 2024;19(1):82.
- Ruiz M, Lacaille F, Berthiller J, Joly P, Dumortier J, Aumar M, et al. Liver disease related to alpha1-antitrypsin deficiency in French children: The DEFI-ALPHA cohort. Liver Int. juin 2019;39(6):1136-46.
- Teckman JH, Rosenthal P, Abel R, Bass LM, Michail S, Murray KF, et Baseline Analysis of a Young a-1-Antitrypsin Deficiency Liver Disease Cohort Reveals Frequent Portal Hypertension. J Pediatr Gastroenterol Nutr. juill 2015;61(1):94-101.
- Fawaz R, Baumann U, Ekong U, Fischler B, Hadzic N, Mack CL, et al. Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. janv 2017;64(1):154-68.
- Sveger Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 10 juin 1976;294(24):1316-21.
- Sveger T, Eriksson The liver in adolescents with alpha 1-antitrypsin deficiency. Hepatology. août 1995;22(2):514-7.
- Hinds R, Hadchouel A, Shanmugham NP, Al-Hussaini A, Chambers S, Cheeseman P, et Variable degree of liver involvement in siblings with PiZZ alpha-1-antitrypsin deficiency-related liver disease. J Pediatr Gastroenterol Nutr. juill 2006;43(1):136-8.
- Joly P, Vignaud H, Di Martino J, Ruiz M, Garin R, Restier L, et ERAD defects and the HFE-H63D variant are associated with increased risk of liver damages in Alpha 1-Antitrypsin Deficiency. PLoS ONE. 2017;12(6):e0179369.
- Joly P, Ruiz M, Garin R, Karatas E, Lachaux A, Restier L, et A Particular SORL1 Micro-haplotype May Prevent Severe Liver Disease in a French Cohort of Alpha 1-Antitrypsin-deficient Children. J Pediatr Gastroenterol Nutr. 1 sept 2021;73(3):e68-72.
- Karatas E, Bouchecareilh Alpha 1-Antitrypsin Deficiency: A Disorder of Proteostasis-Mediated Protein Folding and Trafficking Pathways. Int J Mol Sci. 21 févr 2020;21(4):1493.
- Leon C, Bouchecareilh The Autophagy Pathway: A Critical Route in the Disposal of Alpha 1-Antitrypsin Aggregates That Holds Many Mysteries. Int J Mol Sci. 13 févr 2021;22(4):1875.
- Kamp JC, Kappe NN, Moro CF, Fuge J, Kuehnel MP, Wrenger S, et Fibrosis-Related Gene Profiling in Liver Biopsies of PiZZ a1-Antitrypsin Children with Different Clinical Courses. Int J Mol Sci. 27 janv 2023;24(3):2485.
- Shneider BL, Goodrich NP, Ye W, Sawyers C, Molleston JP, Merion RM, et Nonfasted Liver Stiffness Correlates with Liver Disease Parameters and Portal Hypertension in Pediatric Cholestatic Liver Disease. Hepatol Commun. nov 2020;4(11):1694-707.
- Guillaud O, Dumortier J, Couchonnal-Bedoya E, Ruiz Wilson Disease and Alpha1-Antitrypsin Deficiency: A Review of Non-Invasive Diagnostic Tests. Diagnostics (Basel). 10 janv 2023;13(2):256.
- Francavilla R, Castellaneta SP, Hadzic N, Chambers SM, Portmann B, Tung J, et Prognosis of alpha-1-antitrypsin deficiency-related liver disease in the era of paediatric liver transplantation. J Hepatol. juin 2000;32(6):986-92.
- Guillaud O, Jacquemin E, Couchonnal E, Vanlemmens C, Francoz C, Chouik Y, et Long term results of liver transplantation for alpha-1 antitrypsin deficiency. Dig Liver Dis. mai 2021;53(5):606-11.
- Campbell KM, Arya G, Ryckman FC, Alonso M, Tiao G, Balistreri WF, et High prevalence of alpha-1-antitrypsin heterozygosity in children with chronic liver disease. J Pediatr Gastroenterol Nutr. janv 2007;44(1):99-103.
- Boëlle PY, Debray D, Guillot L, Corvol H, French CF Modifier Gene Study SERPINA1 Z allele is associated with cystic fibrosis liver disease. Genet Med. sept 2019;21(9):2151-5.
- Antonini TM, Girard M, Habes D, Goria O, Debray Optimization of the transition process of youth with liver disease in adulthood: A position paper from FILFOIE, the French network for paediatric and adult rare liver diseases. Clin Res Hepatol Gastroenterol. avr 2020;44(2):135-41.
- Lin HC, Kasi N, Quiros Alpha1-Antitrypsin Deficiency: Transition of Care for the Child With AAT Deficiency into Adulthood. Curr Pediatr Rev. 2019;15(1):53-61.
- Fromme M, Schneider CV, Pereira V, Hamesch K, Pons M, Reichert MC, et Hepatobiliary phenotypes of adults with alpha-1 antitrypsin deficiency. Gut. févr 2022;71(2):415-23.
- Fromme M, Payancé A, Mandorfer M, Thorhauge KH, Pons M, Miravitlles M, et Longitudinal Evaluation of Individuals With Severe Alpha-1 Antitrypsin Deficiency (Pi*ZZ Genotype). Gastroenterology. 15 oct 2024;S0016-5085(24)05572-0.
- Clark VC, Marek G, Liu C, Collinsworth A, Shuster J, Kurtz T, et Clinical and histologic features of adults with alpha-1 antitrypsin deficiency in a non-cirrhotic cohort. J Hepatol. déc 2018;69(6):1357-64.
- Guillaud O, Dumortier J, Traclet J, Restier L, Joly P, Chapuis-Cellier C, et Assessment of liver fibrosis by transient elastography (Fibroscan®) in patients with A1AT deficiency. Clin Res Hepatol Gastroenterol. févr 2019;43(1):77-81.
- Hamesch K, Mandorfer M, Pereira VM, Moeller LS, Pons M, Dolman GE, et Liver Fibrosis and Metabolic Alterations in Adults With alpha- 1-antitrypsin Deficiency Caused by the Pi*ZZ Mutation. Gastroenterology. sept 2019;157(3):705-719.e18.
- Hiller AM, Ekström M, Piitulainen E, Lindberg A, Rönmark E, Tanash Cancer risk in severe alpha-1-antitrypsin deficiency. Eur Respir J. oct 2022;60(4):2103200.
- Strnad P, Buch S, Hamesch K, Fischer J, Rosendahl J, Schmelz R, et Heterozygous carriage of the alpha1-antitrypsin Pi*Z variant increases the risk to develop liver cirrhosis. Gut. juin 2019;68(6):1099-107.
- Ferkingstad E, Oddsson A, Gretarsdottir S, Benonisdottir S, Thorleifsson G, Deaton AM, et Genome-wide association meta-analysis yields 20 loci associated with gallstone disease. Nat Commun. 30 nov 2018;9(1):5101.
- Strnad P, McElvaney NG, Lomas Alpha1-Antitrypsin Deficiency. N Engl J Med. 9 avr 2020;382(15):1443-55.
- Tejwani V, Nowacki AS, Fye E, Sanders C, Stoller The Impact of Delayed Diagnosis of Alpha-1 Antitrypsin Deficiency: The Association Between Diagnostic Delay and Worsened Clinical Status. Respir Care. août 2019;64(8):915-22.
- Miravitlles M, Dirksen A, Ferrarotti I, Koblizek V, Lange P, Mahadeva R, et European Respiratory Society statement: diagnosis and treatment of pulmonary disease in a1-antitrypsin deficiency. Eur Respir J. nov 2017;50(5):1700610.
- Mouzaki M, Bronsky J, Gupte G, Hojsak I, Jahnel J, Pai N, et Nutrition Support of Children With Chronic Liver Diseases: A Joint Position Paper of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. oct 2019;69(4):498-511.
- Lykavieris P, Ducot B, Lachaux A, Dabadie A, Broué P, Sarles J, et Liver disease associated with ZZ alpha1-antitrypsin deficiency and ursodeoxycholic acid therapy in children. J Pediatr Gastroenterol Nutr. nov 2008;47(5):623-9.
- Carey EJ, Iyer VN, Nelson DR, Nguyen JH, Krowka MJ. Outcomes for recipients of liver transplantation for alpha-1-antitrypsin deficiency–related cirrhosis. Liver Transpl. déc 2013;19(12):1370-6.
- Kemmer N, Kaiser T, Zacharias V, Neff Alpha-1-antitrypsin deficiency: outcomes after liver transplantation. Transplant Proc. juin 2008;40(5):1492-4.
- Chapman KR, Burdon JGW, Piitulainen E, Sandhaus RA, Seersholm N, Stocks JM, et Intravenous augmentation treatment and lung density in severe a1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 25 juill 2015;386(9991):360-8.
- Mornex JF, Balduyck M, Bouchecareilh M, Cuvelier A, Epaud R, Kerjouan M, et [French clinical practice guidelines for the diagnosis and management of lung disease with alpha 1-antitrypsin deficiency]. Rev Mal Respir. sept 2022;39(7):633-56.
- Remih K, Amzou S, Strnad Alpha1-antitrypsin deficiency: New therapies on the horizon. Curr Opin Pharmacol. août 2021;59:149-56.
- Loomba R, Clark G, Teckman J, Ajmera V, Behling C, Brantly M, et al. Review article: New developments in biomarkers and clinical drug development in alpha-1 antitrypsin deficiency-related liver disease. Aliment Pharmacol Ther. mai 2024;59(10):1183-95.
- Strnad P, Mandorfer M, Choudhury G, Griffiths W, Trautwein C, Loomba R, et Fazirsiran for Liver Disease Associated with Alpha1- Antitrypsin Deficiency. N Engl J Med. 11 août 2022;387(6):514-24.
- Clark VC, Strange C, Strnad P, Sanchez AJ, Kwo P, Pereira VM, et Fazirsiran for Adults With Alpha-1 Antitrypsin Deficiency Liver Disease: A Phase 2 Placebo Controlled Trial (SEQUOIA). Gastroenterology. 1 oct 2024;167(5):1008-1018.e5.
- Erion DM, Liu LY, Brown CR, Rennard S, Farah Editing Approaches to Treat Alpha-1 Antitrypsin Deficiency. Chest. 12 oct 2024;S0012- 3692(24)05302-9.