Liens d’intérêt
L’auteur déclare n’avoir aucun lien d’intérêt en relation avec cette présentation.
Mots-clés
Kyste du cholédoque ; Anomalies de la plaque ductale ; Polykystose hépatique
Abréviations
AFEF : Association Française pour l’Étude du Foie
AJBP : Anomalie de la jonction bilio-pancréatique
AUDC : Acide ursodésoxycholique
DPC : Duodéno-pancréatectomie céphalique
FHC : Fibrose hépatique congénitale
HTP : Hypertension portale
LPAC : Low phospholipid-associated cholelithiasis
PKHD1 : Polycystic Kidney and Hepatic Disease 1
PKHAD : Polykystose hépatique autosomique dominante
PKRAD : Polykystose rénale autosomique dominante
VBEH : voies biliaires extra-hépatiques
VBIH : voies biliaires intra-hépatiques
VBP : voie biliaire principale
VO : varices œsophagiennes
Introduction et rappels embryologiques sur la formation des canaux biliaires
Les voies biliaires forment un réseau de canaux permettant d’excréter la bile, qui est essentielle à la digestion des graisses et à l’absorption intestinale des lipides, du foie vers l’intestin. On distingue les voies biliaires intra-hépatiques (VBIH) de la voie biliaire principale (VBP) comprenant le canal hépatique commun et le canal cholédoque. Avant son abouchement dans le duodénum, le cholédoque rejoint le canal de Wirsung au niveau de la jonction bilio-pancréatique.
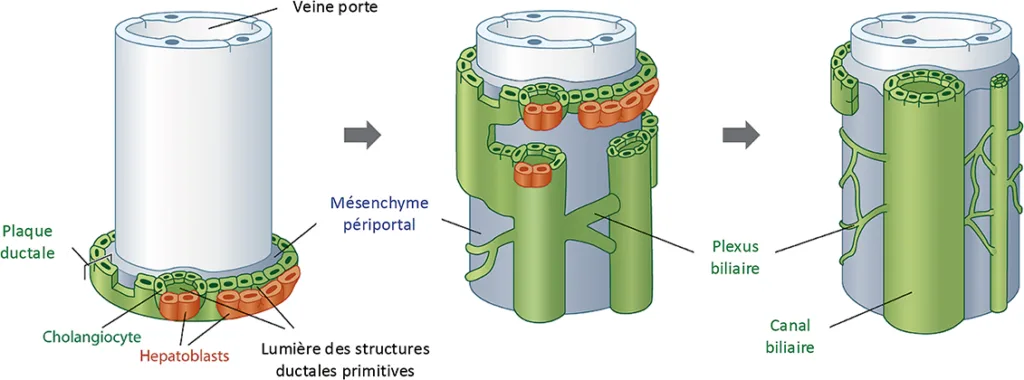
La formation des canaux biliaires intra-hépatiques débute au 2e mois de vie embryonnaire à partir des hépatoblastes issus de l’endoderme du bourgeon hépatique primitif. Certains hépatoblastes vont ainsi s’organiser, autour des branches portales primitives, en une couche monocellulaire appelée plaque ductale (figure 1) (1). La plaque ductale va progressivement se dupliquer, aboutissant à la création d’une lumière entre les 2 couches cellulaires. Cette lumière va focalement se dilater pour former les canaux biliaires primitifs tapissés de cholangiocytes, tandis que le reste des cellules de la plaque ductale va disparaître par apoptose. Les canaux biliaires primitifs vont ensuite s’incorporer dans le mésenchyme périportal pour former l’espace porte définitif. Le développement des canaux biliaires intra-hépatiques débute dans la région du hile hépatique, puis va progressivement s’étendre dans le foie en suivant le développement des branches de la veine porte. Les phénomènes d’organisation et de régression cellulaire de la plaque ductale sont notamment contrôlés par le cil primaire, un organelle non mobile situé à la surface des cellules de la plaque ductale. Le cil primaire, également présent en grande quantité dans le rein, joue un rôle crucial dans la transmission des signaux intracellulaires.
Figure 1 : Formation des canaux biliaires intra-hépatiques à partir de la plaque ductale au cours de l’embryogénèse (1)

À l’extérieur du foie, les canaux biliaires intra-hépatiques se rejoignent au niveau du hile pour former le canal hépatique commun. Les cholangiocytes des voies biliaires extra-hépatiques (VBEH) ont une origine embryologique différente de celle des VBIH : ils proviennent de l’endoderme de la partie ventrale de l’intestin initial (ventral foregut). La vésicule biliaire et le canal cystique dérivent quant à eux du diverticule cystique, lui-même dérivé du bourgeon hépatique primitif (1). La jonction du canal hépatique commun et du canal cystique permet la formation du canal cholédoque. La jonction bilio-pancréatique résulte de la fusion de la VBP primitive avec les canaux du pancréas ventral et dorsal, à la partie postérieure du duodénum.
Au cours de l’embryogénèse, des anomalies de développement des VBIH ou de la VBP peuvent survenir, entraînant des malformations congénitales de présentation et de gravité variable. Nous détaillerons les principales malformations des canaux biliaires ainsi que les variations anatomiques les plus fréquemment retrouvées en pratique clinique. L’atrésie des voies biliaires, pathologie essentiellement pédiatrique, ne sera pas détaillée.
Principales variations anatomiques d’intérêt
Variations anatomiques de la vésicule biliaire
L’agénésie de la vésicule biliaire et du canal cystique est causée par l’absence de développement du diverticule cystique. C’est une anomalie rare, avec une incidence estimée entre 10 et 65 pour 100 000 (2). L’agénésie vésiculaire est le plus souvent isolée (70 % des cas), mais elle peut parfois s’intégrer dans un syndrome polymalformatif associant malformations ventriculaires et imperforations anales. Une agénésie vésiculaire se présente dans plus de 50 % des cas sous la forme d’épisodes récidivants de colique hépatique, possiblement liés à une dysfonction du sphincter d’Oddi ou à une lithiase de la voie biliaire principale. La réalisation d’une échographie hépatique puis d’une cholangio-IRM est nécessaire pour éliminer la présence d’une vésicule biliaire ectopique. En cas de lithiase de la VBP, l’extraction du calcul par CPRE est nécessaire. En cas de douleurs persistantes sans lithiase, associées à une dysfonction du sphincter d’Oddi, la prescription de myorelaxants peut être utile. Dans de rare cas, une sphinctérotomie peut être nécessaire, mais le risque élevé de pancréatite (> 30 %) doit être clairement expliqué au patient (2).
Figure 2 : Aspect en cholangio-IRM de duplication vésiculaire, avec présence d’un macrocalcul dans la vésicule antérieure (31)

La duplication vésiculaire, due à un défaut de vacuolisation du diverticule cystique, peut se présenter sous la forme de 2 vésicules distinctes avec 2 canaux cystiques s’abouchant dans le cholédoque, ou sous la forme d’une vésicule principale associée à une vésicule accessoire de plus petite taille (figure 2) (3). Sa prévalence est estimée à 1/4 000 naissances. La duplication vésiculaire expose au risque de complication chirurgicale en cas de mauvaise évaluation anatomique préopératoire. La persistance d’un diverticule « infundibulaire » (ou diverticule de Hartmann) peut parfois entraîner une compression de la voie biliaire principale.
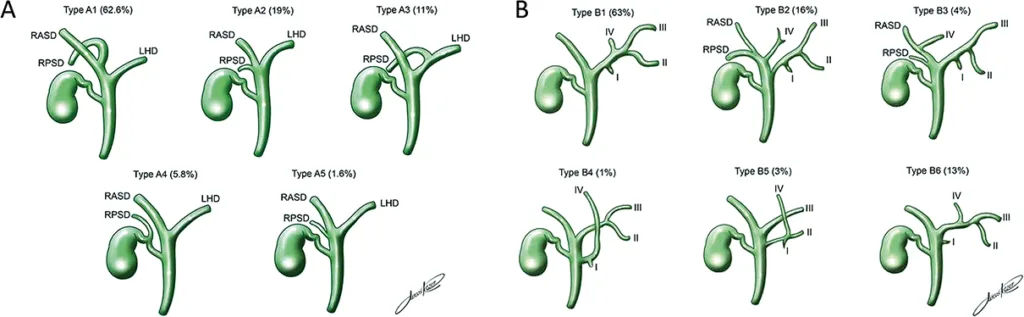
Figure 3 : Classification de Huang et fréquence des variations anatomiques des canaux biliaires intra-hépatiques droits (A) et gauches (B) (4)

Variations anatomiques des voies biliaires intra-hépatiques et de la voie biliaire principale
La présentation modale des VBIH et de la VBP concerne 75 % de la population générale. Il existe de nombreuses variations anatomiques des VBIH, principalement par variation de l’abouchement des canaux biliaires sectoriels dans le canal hépatique droit ou gauche (figure 3) (4). Ces variantes anatomiques sont le plus souvent asymptomatiques mais peuvent être à l’origine de difficultés techniques en cas de cathétérisme biliaire. Dans ces conditions, une cartographie des voies biliaires par cholangio-IRM est souvent nécessaire avant la réalisation du geste endoscopique.
Les principales variantes anatomiques de la VBP rencontrées à l’étage supérieure sont (5) :
- Une trifurcation biliaire au niveau de la convergence biliaire due à une agénésie du canal hépatique droit, avec présence de 2 canaux biliaires droits (antérieur et postérieur) reliés directement au canal hépatique gauche pour former le canal hépatique commun (figure 4A) ;
- Une convergence biliaire étagée avec abouchement d’un canal hépatique sectoriel (le plus souvent du foie droit) directement dans la voie biliaire principale (figure 4B) ;
- Un canal hépato-cystique (due à un défaut de séparation entre le bourgeon hépatique et le bourgeon cystique) assurant le drainage biliaire d’un secteur hépatique droit vers la vésicule biliaire ou le canal cystique. Leur fréquence est évaluée à 5 % de la population générale ;
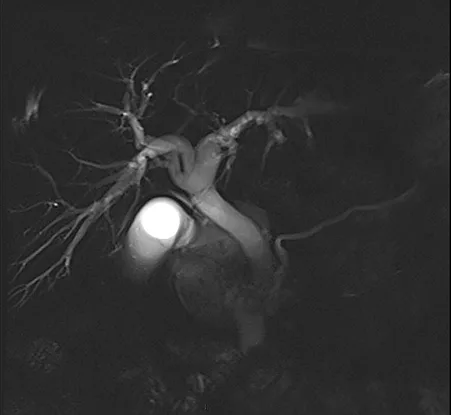
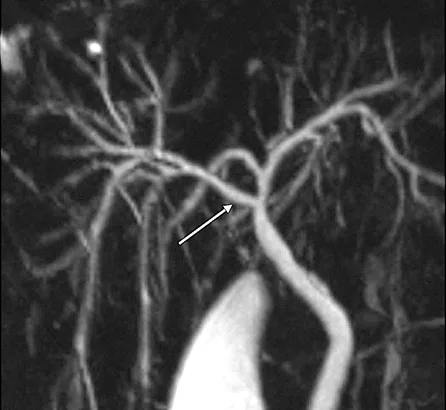
Figure 4 : Aspect en cholangio-IRM des principales variations anatomiques des voies biliaires : trifurcation biliaire (A) ; convergence biliaire étagée avec abouchement direct du canal hépatique antérieur droit (flèche) dans le cholédoque (B) ; canal de Luschka (flèche) compliqué de biliome (étoile) (C) ; gros cholédoque (D)




- Le canal de « Luschka » ou canal biliaire sous-vésiculaire, est un canal biliaire aberrant naissant en dehors du parenchyme hépatique, le plus souvent dans l’espace périvésiculaire, et se drainant dans un canal biliaire intra-hépatique, majoritairement du foie droit.
Ces variations anatomiques sont généralement asymptomatiques mais exposent au risque, en cas de cholécystectomie ou de chirurgie hépatique, de plaie biliaire ou de fuite biliaire dans le cas du canal de « Luschka » (figure 4C). La cholangio-IRM pré-opératoire permet le plus souvent de repérer ces variations et de prévenir une éventuelle complication chirurgicale. Cependant, il n’existe pas de donnée justifiant la réalisation systématique d’une cholangio-IRM avant chirurgie vésiculaire, et celle-ci n’est donc pas recommandée.
Le cholédoque peut être également le siège de variations anatomiques. La principale est une dilatation modérée du cholédoque ou « gros cholédoque », que l’on peut retrouver après cholécystectomie ou chez la personne âgée (figure 4D). Devant un gros cholédoque, l’essentiel est d’éliminer un obstacle de la VBP (sténose tumorale ou inflammatoire, calcul) et une anomalie de la jonction bilio-pancréatique (cf. infra).
La duplication du cholédoque est un évènement rare, et est souvent associée à des anomalies de la jonction bilio-pancréatique. Le principal diagnostic différentiel est la présence d’un canal cystique long s’abouchant dans le bas cholédoque et pouvant être confondu avec un 2e cholédoque.
Kystes du cholédoque et anomalies de la jonction bilio-pancréatique
Les kystes du cholédoque sont des malformations congénitales rares des voies biliaires caractérisées par une dilatation des voies biliaires intra et/ou extra-hépatiques. Il s’agit de la 2e cause de malformations congénitales des voies biliaires après l’atrésie des voies biliaires. L’incidence est nettement plus élevée dans les populations asiatiques (1/13 000) que dans les populations occidentales (1/150 000), mais aucun facteur étiologique expliquant cette différence n’a encore été identifié (6). On retrouve une nette prédominance féminine avec un sex ratio de 4 femmes pour 1 homme.
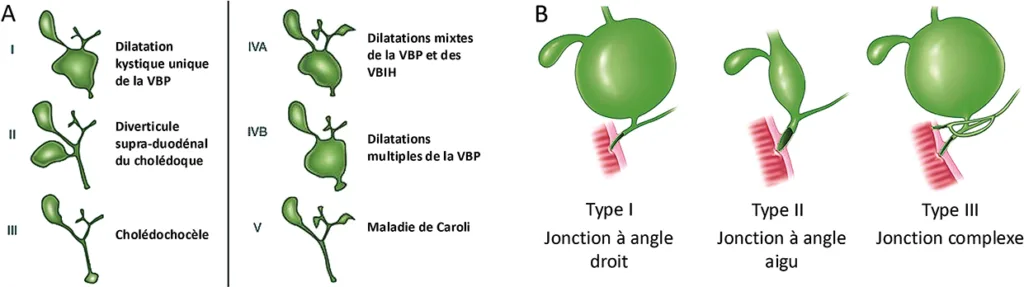
Il existe plusieurs classifications morphologiques des kystes du cholédoque, basées sur l’aspect en cholangiographie. La plus couramment utilisée est celle de Todani qui distingue les kystes du cholédoque en 5 classes (figure 5A) :
- Type I : dilatation kystique unique de la VBP
- Type II : diverticule supra-duodénal du cholédoque
- Type III : dilatation kystique de la portion terminale du cholédoque (cholédochocèle)
- Type IV divisé en 2 sous-types :
- IVa : dilatations kystiques mixtes de la VBP et des VBIH
- IVb : dilatations kystiques multiples de la VBP
- Type V : dilatations kystiques des VBIH, correspondant à la maladie de Caroli (cf. infra)
Les types I et IVa sont les plus fréquemment décrits (> 80 % des cas), avec une prédominance de type I dans les formes de révélation pédiatrique, et une prédominance de type IVa chez les adultes (6).
La principale cause identifiée de kystes du cholédoque est l’anomalie de la jonction bilio-pancréatique (AJPB), retrouvée chez plus de 90 % des patients avec dilatation kystique du cholédoque. L’AJPB résulte d’une anomalie de fusion de la voie biliaire principale primitive avec les canaux du pancréas ventral et du pancréas dorsal. L’AJPB est définie, selon les recommandations japonaises, par la présence d’un canal commun bilio- pancréatique de longueur anormale (> 10 mm) associé à une jonction bilio-pancréatique située en dehors de la paroi duodénale (7). Un taux anormal d’enzymes pancréatiques dans les voies biliaires, mesuré par cathétérisme biliaire, peut également conduire au diagnostic d’AJBP en cas de doute sur l’imagerie. L’AJBP est responsable d’un reflux des enzymes pancréatiques dans le cholédoque, entraînant une augmentation de la pression intra-biliaire, une fragilisation de l’épithélium biliaire puis une dilatation du cholédoque. On distingue 3 types d’AJPB selon le type et l’angle de la jonction entre les canaux biliaires et pancréatiques (classification de Komi, figure 5B).
Figure 5 : Classification de Todani des kystes du cholédoque (A), classification de Komi des anomalies de la jonction bilio- pancréatique (B) (32, 33)

Les kystes du cholédoque sont diagnostiqués, dans plus de 80 % des cas, dans l’enfance car la symptomatologie est souvent bruyante, sous la forme d’un ictère associé à des douleurs abdominales et une hépatomégalie (8). Cependant, il existe des formes de diagnostic plus tardif, à l’âge adulte, dont l’expression clinique peut être moins spécifique, et se limiter à des douleurs de l’hypochondre droit (9). Les kystes du cholédoque peuvent également être diagnostiqués dans les suites d’une complication biliaire lithiasique à type de cholécystite ou d’angiocholite, suite à un abcès hépatique ou bien d’emblée au stade de cirrhose biliaire secondaire avec hypertension portale (8). La péritonite biliaire par rupture de kyste est une complication grave mais rare (< 2 %), qui peut survenir classiquement en cours de grossesse (10). Il existe également une forte prévalence de pancréatites aiguës (70 %) dans cette population, dont l’origine peut être lithiasique ou bien liée au reflux biliaire dans le canal pancréatique principal (8). L’expression clinique des kystes du cholédoque peut différer selon leur morphologie. Ainsi, les kystes de type II sont le plus souvent asymptomatiques, tandis que les kystes de type III se révèlent plus fréquemment par un épisode de pancréatite aiguë ou par un syndrome occlusif haut par obstruction de la lumière duodénale. Les kystes de type IVa et V semblent être les plus associés aux complications biliaires lithiasiques.
La survenue de cancers des voies biliaires (cholangiocarcinome et adénocarcinome de la vésicule biliaire) constitue la complication la plus grave des kystes du cholédoque. L’incidence globale des cancers des voies biliaires dans cette population est évaluée à 10,7 % (11). Cependant, l’incidence varie en fonction de l’âge : elle est très faible dans l’enfance (< 1 %) et augmente progressivement avec l’âge pour atteindre 40 % après 60 ans (12). L’âge médian au diagnostic est de 49 ans. L’inflammation chronique de l’épithélium biliaire résultant du reflux des enzymes pancréatiques dans les voies biliaires semble être le principal facteur étiologique expliquant l’incidence élevée des cancers chez ces patients. Les kystes de type I et IV sont les plus à risque de dégénérescence et regroupent 90 % des cas de cancer (11). En cas d’AJBP sans kyste du cholédoque associé, le risque de cancer est concentré sur la vésicule biliaire avec une incidence globale supérieure à 40 % (7).
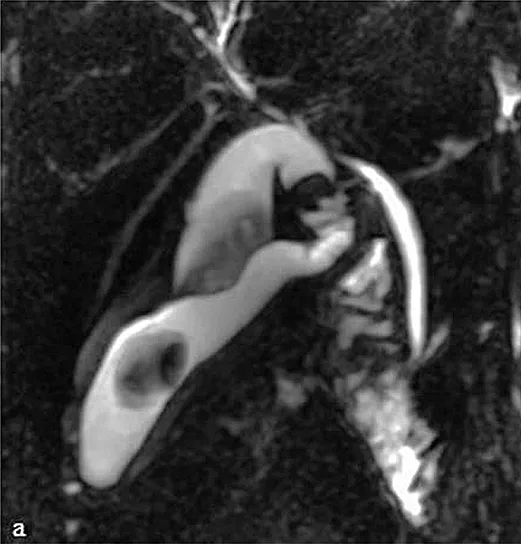
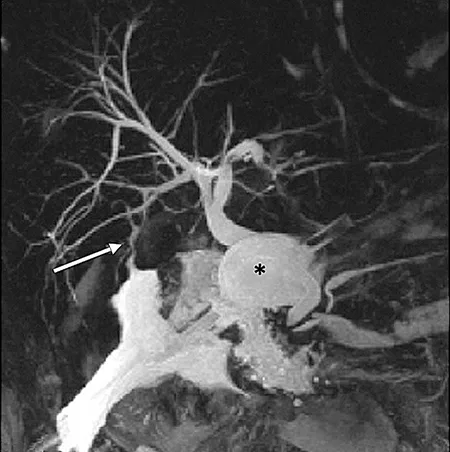
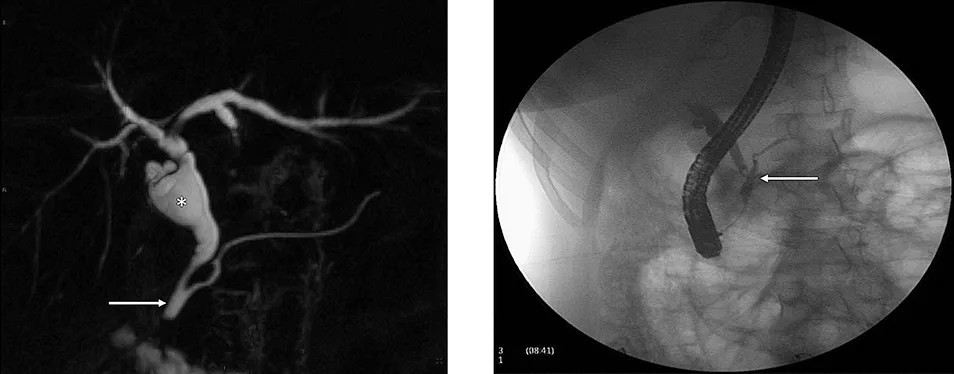
Figure 6 : Aspect en cholangio-IRM d’un kyste du cholédoque de type I (étoile) associé à une anomalie de jonction bilio-pancréatique de type II (flèche) (A). Clichés cholangiographiques obtenus par CPRE d’une anomalie de jonction bilio-pancréatique de type I (B)

La cholangio-IRM est l’examen de référence pour le diagnostic de kyste du cholédoque (figure 6A). La cholangio-IRM permet de confirmer le diagnostic par la visualisation d’une communication entre la dilatation kystique et les voies biliaires et par l’élimination des principaux diagnostics différentiels, notamment des kystes extra-biliaires non communiquants. L’IRM hépatique permet également l’évaluation du retentissement du kyste sur le parenchyme hépatique (dysmorphie hépatique, abcès) et le dépistage des complications (cancers, hypertension portale). La visualisation d’une AJBP en IRM est essentielle pour distinguer un kyste du cholédoque d’un gros cholédoque du sujet âgé ou secondaire à une cholécystectomie (cf. supra). En cas de doute sur l’existence d’une AJBP qui peut être de visualisation difficile sur la cholangio-IRM, l’écho-endoscopie bilio-pancréatique permet généralement de confirmer le diagnostic. Compte tenu du risque important d’angiocholite, la réalisation d’une CPRE diagnostique (figure 6B) doit être réservée aux suspicions de cholangiocarcinome afin de réaliser des prélèvements histologiques.
La prise en charge des kystes du cholédoque diffère selon le type de kyste. La résection totale du kyste (donc de la VBP) avec anastomose hépatico- jéjunale sur anse en Y est le traitement recommandé par l’Association Française pour l’Étude du Foie (AFEF) pour les kystes de type I et IV (13). La cholécystectomie doit également être systématiquement réalisée dans le même temps. Compte tenu de la morbidité de la chirurgie pancréatique, il n’est pas conseillé de réaliser l’exérèse du cholédoque intrapancréatique par duodéno-pancréatectomie céphalique (DPC) en l’absence de signe de cholangiocarcinome du bas cholédoque. De même, pour les kystes de type IVa avec atteinte unilatérale des VBIH, une hépatectomie partielle n’est pas systématique mais peut être proposée en cas de lithiase intrahépatique compliquée d’angiocholites à répétition (avec calcul non extractible par endoscopie) ou en cas de cholangiocarcinome intra-hépatique. Les complications à court terme de la chirurgie peuvent être pancréatiques (fistules, pancréatite aigüe) ou biliaires (fistules anastomotiques, péritonites biliaires) (14). Les complications à long terme de la chirurgie d’exérèse, de l’ordre de 15 %, sont dominées par les sténoses de l’anastomose biliaire et les complications lithiasiques, notamment pour les kystes de type IV (15). Dans de rares cas (< 10 %), une reprise chirurgicale peut être nécessaire pour réfection de l’anastomose bilio-digestive ou exérèse de dilatations kystiques résiduelles (15). Bien que la chirurgie d’exérèse permette de diminuer considérablement le risque de dégénérescence de l’épithélium biliaire, il persiste un risque de cancer. Dans une série rétrospective japonaise de 94 patients, 4 patients développaient un cholangiocarcinome à distance de la chirurgie, avec une incidence cumulée de 11 % à 25 ans (16). Dans cette série, tous les cancers survenaient sur des segments biliaires non réséqués (intra-hépatique, hilaire, intra-pancréatique).
Pour les kystes de type II et III, compte tenu du faible risque de cholangiocarcinome, la résection complète de la VBP n’est pas nécessaire. L’exérèse du diverticule biliaire est suffisante pour les kystes de type II (8). Pour les kystes de type III, une sphinctérotomie endoscopique est souvent proposée afin de prévenir le risque de lithiase obstructive de la VBP (8). En cas d’AJBP associée, il reste cependant justifié de réaliser une cholécystectomie préventive du fait du risque élevé de cancer vésiculaire.
Il n’existe pas de recommandation détaillant la surveillance des kystes du cholédoque opérés. Néanmoins, compte tenu du risque de complications post-opératoires notamment lithiasiques et du risque de cancer résiduel, une surveillance annuelle par cholangio-IRM doit être maintenue.
Lésions kystiques des voies biliaires intra-hépatiques liées à une malformation de la plaque ductale
En cas d’anomalie de structure ou de fonctionnement du cil primaire (ciliopathie), le développement et la réorganisation de la plaque ductale au cours de l’embryogénèse peut être altérée et aboutir à des anomalies des voies biliaires. La plupart des ciliopathies sont en lien avec des anomalies génétiques. L’expression clinique des malformations biliaires associées à une anomalie de la plaque ductale est différente suivant la période de l’embryogénèse concernée par l’anomalie. On distingue ainsi 3 grands syndromes : la maladie de Caroli, atteignant les canaux biliaires segmentaires (atteinte précoce), la fibrose hépatique congénitale (FHC) atteignant les canaux biliaires interlobulaires (atteinte plus tardive) et les hamartomes biliaires intéressant les canaux biliaires périphériques (atteinte tardive). Ces différents tableaux peuvent cependant s’associer pour former des atteintes plus complexes.
Maladie de Caroli et fibrose hépatique congénitale
La maladie de Caroli est un syndrome malformatif des VBIH définit par une dilatation kystique des canaux biliaires segmentaires. Occasionnellement, des anomalies des VBEH à type de dilatation peuvent s’associer. Dans la majorité des cas, cette dilatation kystique des VBIH est associée à une FHC, on parle alors de syndrome de Caroli (tableau 1). La FHC se caractérise par la présence dans un espace porte fibreux de nombreux canaux biliaires ectasiques, communiquant avec le reste de l’arbre biliaire, et parfois dilatés formant alors des kystes microscopiques. Fréquemment, le syndrome de Caroli est associé à des malformations rénales, de type ectasies des tubules rénaux voire polykystose rénale.
La maladie de Caroli isolée est le plus souvent d’origine sporadique. À l’inverse, le syndrome de Caroli est associée dans la majorité des cas à des mutations du gène Polycystic Kidney and Hepatic Disease 1 (PKHD1) qui code pour la fibrocystine, une protéine impliquée dans le fonctionnement du cil primaire (tableau 2) (17). Les mutations de PKHD1 sont transmises selon un mode autosomique récessif. Il existe une corrélation entre le génotype et le phénotype associé aux mutations de PKHD1 : la présence de variant non-sens bi-alléliques, responsable d’une protéine tronquée, est associée à un phénotype plus sévère que des variants faux-sens (17).
La prévalence de la polykystose rénale autosomique récessive associée à une FHC est de l’ordre de 1/100 000 naissances (18). La maladie de Caroli isolée est plus rare avec une prévalence estimée à 1/1 000 000 naissances (19).
La maladie de Caroli est le plus souvent diagnostiquée chez l’adolescent ou l’adulte jeune, à la suite d’épisodes répétés d’angiocholites, parfois compliqués d’abcédation du foie. Dans la forme typique, le diagnostic d’angiocholite est souvent difficile car les douleurs biliaires et l’ictère sont absents en l’absence de migration de calculs dans la voie biliaire principale (20). La survenue d’angiocholite est plus fréquente dans le syndrome de Caroli que dans la maladie de Caroli isolée (tableau 1). La survenue d’une angiocholite est un évènement grave car responsable d’un 1/3 des décès (21). La FHC se caractérise par la présence d’une hypertension portale (HTP) sans cirrhose, et le mode de révélation le plus fréquent est la rupture de varices œsophagiennes (VO), qui peut survenir dès l’enfance ou chez l’adulte jeune. Tous comme les kystes du cholédoque, la maladie de Caroli constitue un facteur de risque important de cholangiocarcinome. L’incidence du cholangiocarcinome dans cette population est ainsi estimée à 6,5 %, avec un risque relatif 38 fois supérieur à la population générale (22).
Tableau 1 : Caractéristiques cliniques de la maladie de Caroli isolée et du syndrome de Caroli associé à la fibrose hépatique congénitale (20)
| Maladie de Caroli isolée | Syndrome de Caroli |
| Transmission | Sporadique | Autosomique récessif |
| Distribution des kystes | Diffuse ou localisée | Diffuse |
| Angiocholite | Possible | Fréquente |
| Hypertension portale | Absente | Fréquente |
| Anomalies rénales | Absente | Ectasies tubulaires Polykystose |
Tableau 2 : Mode de transmission et caractérisation génétique des principales malformations des voies biliaires associées à une anomalie de plaque ductale
| Transmission | Gènes | Protéine |
| Maladie de Caroli | Sporadique (dans la majorité des cas) | | |
| Syndrome de Caroli | AR | PKHD1 | Fibrocystine |
| Hamartomes | Sporadique (dans la majorité des cas) | | |
| PKRAD | AD | PKD1 PKD2 | Polycystine-1 Polycystine-2 |
| PKHAD | AD | PRKCSH Sec63 | Hépatocystine Réticulum endoplasmique |
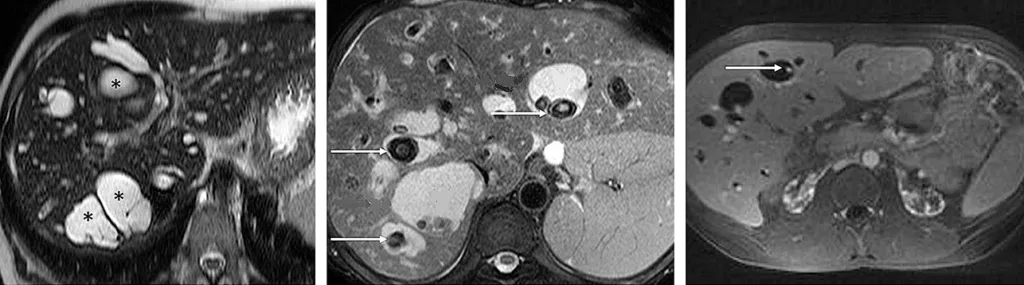
Le diagnostic positif de maladie de Caroli repose sur la cholangio-IRM. Celle-ci met en évidence de volumineuses dilatations kystiques des VBIH, en communication avec le reste de l’arbre biliaire (figure 7A). Il est fréquent de visualiser des calculs au sein des dilatations kystiques (figure 7B). Après injection de produit de contraste, il est classique de visualiser un vaisseau réhaussé (branche de la veine porte ou de l’artère hépatique) au sein de la dilatation kystique (figure 7C). Ce signe radiologique dit « central du point » (central dot sign), témoin d’une malformation de la plaque ductale, est pathognomique de la maladie de Caroli. Dans la maladie de Caroli isolée, les dilatations kystiques peuvent être localisées au niveau d’un lobe ou d’un segment hépatique, alors qu’elles sont généralement diffuses dans le cadre du syndrome de Caroli (tableau 1). Concernant le diagnostic de FHC, l’apport de l’imagerie est plus limité, mais permet d’évaluer la présence de signes radiologiques d’HTP (splénomégalie, voies de dérivations porto-systémiques). Il peut néanmoins exister des lésions biliaires associées à la FHC isolée sans syndrome de Caroli, à type de cholangite ou d’augmentation de volume de la vésicule biliaire (23).
Figure 7 : Visualisation par IRM hépatique de lésions caractéristiques de maladie de Caroli : dilatation kystiques (étoiles) des canaux biliaires intra-hépatiques (A) ; présence de calculs (flèches) au sein des dilatations kystiques biliaires (B) ; central dot sign : vaisseau réhaussé au sein d’une dilatation kystique (flèche) (C)

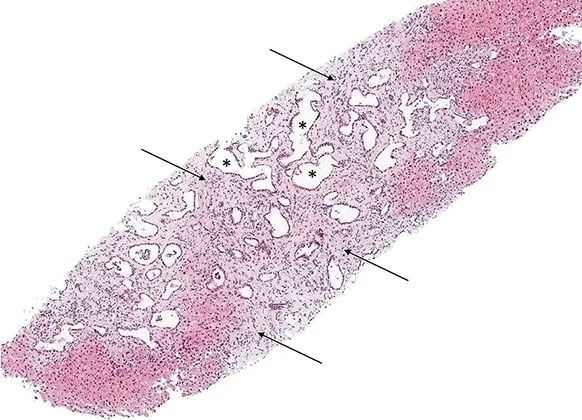
Figure 8 : Coupe histologique (coloration hématoxyline- éosine) d’une biopsie hépatique d’un patient atteint de fibrose hépatique congénitale : volumineux canaux biliaires ectasiques (étoiles) situés au sein d’une fibrose septale dense (flèches)

Contrairement à la maladie de Caroli, la biopsie hépatique est indispensable au diagnostic de FHC. Classiquement, l’histologie retrouve une fibrose septale dense avec présence en son sein de nombreux canaux biliaires de grande taille, ectasiques, communiquant avec le reste des voies biliaires (figure 8). En cas de syndrome de Caroli, les canaux biliaires ectasiques interlobulaires peuvent confluer en structures kystiques (17). Il existe également des anomalies vasculaires avec des veinules portes hypoplasiques. La préservation de l’architecture globale du lobule et l’absence de fibrose mutilante permet d’éliminer une cirrhose, qui reste le principal diagnostic différentiel histologique.
La prise en charge de la maladie de Caroli repose sur le traitement des angiocholites. Une antibiothérapie, adaptée si possible à une documentation bactériologique, doit être systématiquement instaurée. Le traitement des calculs est également essentiel pour prévenir les angiocholites. L’acide ursodésoxycholique (AUDC) est généralement proposé dans la maladie de Caroli. Grâce à son effet cholérétique, l’AUDC pourrait prévenir l’apparition de calculs et diminuer le risque d’angiocholite (24). En cas de calcul symptomatique enclavé dans la VBP, la CPRE peut être discutée, mais le rapport bénéfice-risque doit être évalué précisément compte tenu du risque important d’angiocholite. La prise en charge de l’HTP secondaire à la FHC n’a pas été discutée dans la dernière conférence de consensus BAVENO VII consacrée au dépistage et au traitement de l’HTP. Néanmoins, on peut proposer de traiter l’HTP selon les mêmes modalités que pour l’HTP secondaire à la cirrhose : ligature endoscopique des VO ou sclérothérapie des varices gastriques en cas de saignement, associé à un traitement par bêta-bloquant non cardio-sélectif (carvedilol ou propranolol) au long cours. Le TIPS est réalisé chez 20 % des patients avec FHC, avec des bons résultats en cas d’HTP incontrôlée. Cependant, les patients doivent être sélectionnés rigoureusement car le risque d’encéphalopathie peut être important en cas de maladie hépatique et/ou rénale avancée (21). Une hépatectomie partielle est parfois envisagée en cas de maladie de Caroli localisée et symptomatique, sans hypertension portale. La transplantation hépatique (+/- combinée avec la transplantation rénale) reste le traitement de dernier recours en cas d’angiocholite à répétition sans possibilité de traitement chirurgical ou en cas d’hypertension portale décompensée (25).
Le risque de cholangiocarcinome dans la maladie de Caroli justifie une surveillance rapprochée. L’AFEF recommande de dépister tous les patients atteints de maladie de Caroli par une IRM hépatique annuelle (13). Concernant le dépistage et la surveillance de l’HTP associée à la FHC, en l’absence de recommandation, la réalisation d’une gastroscopie régulière pour dépister les VO apparaît licite.
Hamartomes biliaires (complexes de von Meyenburg)
Les hamartomes biliaires sont des structures biliaires de petite taille, résultant d’une malformation tardive de la plaque ductale et atteignant les canaux biliaires les plus périphériques. L’étiologie des hamartomes est encore discutée. Bien que dans la majorité des cas, ils soient d’origine sporadique, l’association d’hamartomes biliaires avec des mutations hétérozygotes du gène PKHD1 a déjà été rapporté chez les parents de patients atteints de polykystose rénale autosomique récessive (tableau 2) (26). La dilatation progressive des hamartomes est également suspectée d’être à l’origine de la polykystose hépatique associée à la polykystose rénale autosomique dominante (cf. infra).
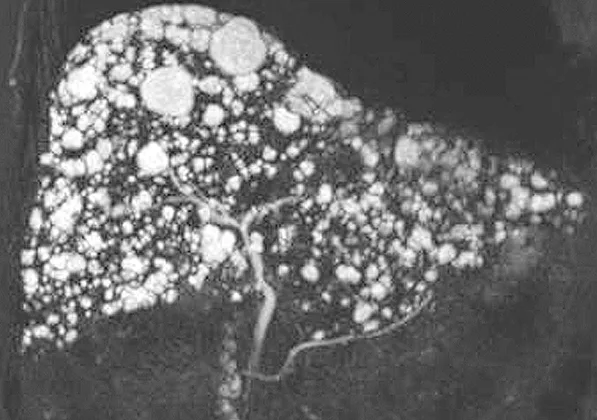
Les hamartomes biliaires sont habituellement asymptomatiques et de découverte fortuite. Bien qu’ils puissent être associés à un syndrome de Caroli ou à une FHC, ils sont le plus souvent identifiés sur un foie normal. En échographie hépatique, les hamartomes peuvent apparaître hypo ou anéchogène, mais sont parfois visualisés sous la forme d’artéfacts en « queue de comète », ce qui pose le problème du diagnostic différentiel avec la microlithiase du syndrome LPAC (Low phospholipid-associated cholelithiasis) (27). En cholangio-IRM, les hamartomes apparaissent sous la forme de lésions kystiques en hyposignal T1 et hypersignal T2, de taille et de nombre variable, localisées en situation péri-portale (28). Ces lésions ne sont pas communicantes avec les voies biliaires, ce qui permet de les différencier d’une maladie de Caroli. En cas de lésions disséminées dans l’ensemble du foie, on parle parfois de foie en « ciel étoilé » (figure 9).
Figure 9 : Aspect en cholangio-IRM de multiples hamartomes biliaires réalisant un aspect de foie « en ciel étoilé »

L’histologique hépatique n’est pas nécessaire au diagnostic d’hamartome biliaire mais ils sont parfois diagnostiqués fortuitement sur une biopsie réalisée pour une autre indication ou sur une pièce d’hépatectomie. L’hamartome se présente habituellement sous la forme de petits canaux biliaires irréguliers et dilatés, non communicants, entourés d’un tissu fibreux (17).
Le rôle des hamartomes biliaires dans le développement des cholangiocarcinomes est débattu. Certaines observations ont rapporté de rare cas de dégénérescence d’hamartomes biliaires en cholangiocarcinome intra-hépatique mais en l’absence de donnée robuste, l’AFEF ne recommande pas de dépistage systématique des patients (13). Il n’y a donc aucune prise en charge spécifique à proposer en cas de diagnostic d’hamartomes biliaires et l’absence thérapeutique reste la règle dans cette situation.
Polykystose hépatique et rénale autosomique dominante
La polykystose hépatique associée à la polykystose rénale autosomique dominante (PKRAD) est la plus fréquente des ciliopathies. La prévalence de la PKRAD est de 1 pour 2 500 habitants en Europe (29). Dans la majorité des cas, elle est secondaire à des mutations du gène PKD1 (80 %) ou PKD2 (15 %) codant respectivement pour les protéines polycystine-1 et polycystine-2 impliquées dans la fonction du cil primaire (tableau 2). Bien que la transmission soit autosomique dominante, la survenue d’une 2e mutation somatique (en plus de la mutation germinale transmise) est nécessaire à l’apparition de la maladie. Au-delà des facteurs génétiques, il semble que les œstrogènes soient également impliqués dans la formation et le développement des kystes. En effet, la PKRAD est plus fréquente chez les femmes (80 % de femmes), et il a été montré que le nombre et la taille des kystes augmentait avec l’exposition aux œstrogènes (grossesses, contraception œstro-progestative, traitement hormonal substitutif de la ménopause) (28).
Sur un plan clinique, l’apparition des kystes hépatiques est toujours retardée par rapport aux kystes rénaux et leur prévalence augmente progressivement avec l’âge. Elle est ainsi de 0 % à 20 ans et atteint 80 % à 60 ans (20). Dans la plupart des cas, la polykystose hépatique est asymptomatique et contrairement au rein, la fonction hépatique est toujours conservée. En cas de kystes volumineux, les symptômes sont liés majoritairement à la compression du foie sur les organes adjacents : douleurs abdominales, dyspnée d’effort, vomissements et satiété précoce, dénutrition. Plus rarement, les kystes peuvent se compliquer d’hémorragie ou d’infection intra-kystique, voire de rupture dans la cavité abdominale. Dans 10 % des cas, la PKRAD est associée à des malformations vasculaires, principalement des anévrysmes cérébraux.
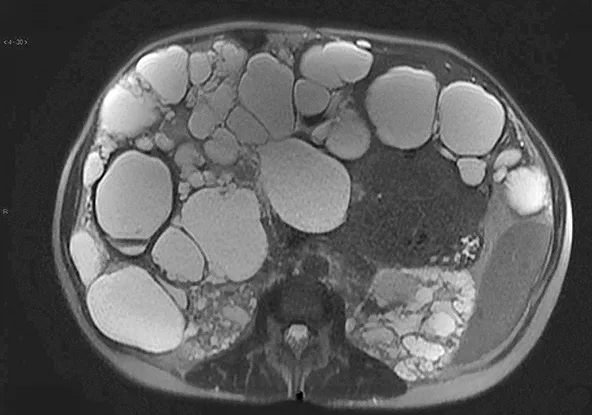
Figure 10 : Polykystose hépatique secondaire à une mutation de PKD1 visualisée sur une IRM hépatique en séquence pondérée T2

Le diagnostic est facilement porté par l’échographie hépatique qui montre de multiples kystes (> 10) supracentimétriques, au contenu anéchogène, avec renforcement postérieur. Ces kystes ne sont pas communicants avec les voies biliaires. En cas de polykystose hépatique non compliquée, le scanner ou l’IRM hépatique ne sont pas indiqués. L’IRM montre classiquement de volumineux kystes non communiquant, en hyposignal T1 et hypersignal T2 (figure 10). La présence de bulles d’air intra-kystiques permet de poser le diagnostic d’infection de kyste. L’hémorragie intra-kystique peut être diagnostiquée en échographie par la visualisation d’un sédiment intra-kystique et en IRM par la visualisation d’un contenu intra-kystique hétérogène en hypersignal T1 et T2 qui témoigne du saignement récent.
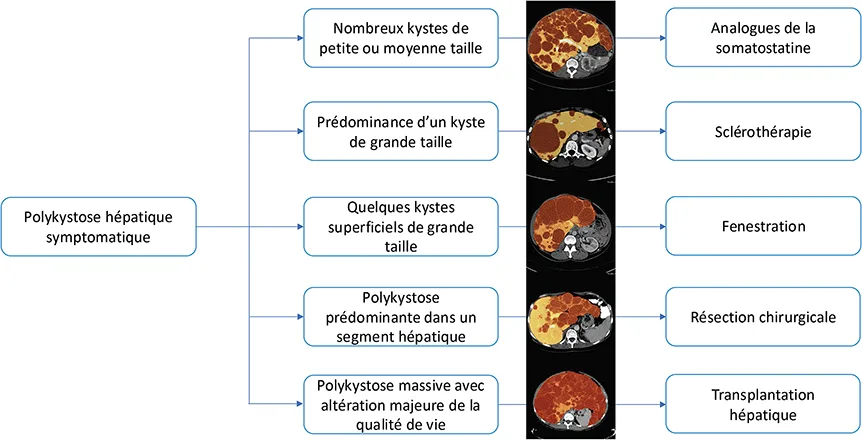
Figure 11 : Stratégie thérapeutique de la polykystose hépatique symptomatique (28)

La polykystose hépatique, en l’absence de symptôme, ne justifie aucun traitement ni suivi particulier. En cas de polykystose symptomatique, il existe différentes modalités thérapeutiques dont l’indication va dépendre du nombre et de la taille des kystes hépatiques. Ces différents traitements ont été discutés dans de récentes recommandations européennes (figure 11) (28). De manière générale, les traitements hormonaux à base d’œstrogènes doivent être contre-indiqués. Les analogues de la somatostatine ont montré une efficacité sur la réduction du volume des kystes et sur l’amélioration de la qualité de vie des patients avec un effet pouvant être maintenu jusqu’à 4 ans après le début du traitement. La sclérothérapie, réalisée par voie radiologique, permet de traiter un kyste symptomatique de grande taille mais la récidive des symptômes après traitement est fréquente. Plusieurs techniques chirurgicales ont été évaluées dans la polykystose hépatique symptomatique. La fenestration des kystes peut être réalisée en présence de kystes de grande taille, peu nombreux, et situés en périphérie du foie. Cette technique est efficace mais elle pose le problème d’adhérences post- opératoires qui peuvent compliquer un geste ultérieur de résection ou de greffe hépatique. En cas de polykystose majoritairement localisée dans un lobe ou segment hépatique, la résection chirurgicale peut être discutée. Compte tenu du risque important de complications post-opératoires (ascite, hémorragie, pleurascite) et d’une mortalité non négligeable (2 %), la résection chirurgicale ne doit s’envisager qu’en cas de contre-indication à la transplantation hépatique. La transplantation hépatique est proposée en cas de polykystose hépatique massive non accessible aux traitements précédemment décrits, avec altération très importante de la qualité de vie. Elle peut être associée à la transplantation rénale en cas d’insuffisance rénale chronique sévère (clairance de la créatinine < 30 mL/min).
La polykystose hépatique autosomique dominante sans polykystose rénale (PKHAD) est le principal diagnostic différentiel de la PKRAD. Elle est associée le plus souvent à des mutations des gènes PRKCSH et Sec63, impliqués dans la structure du réticulum endoplasmique nécessaire au fonctionnement du complexe de polycystines (tableau 2) (30). La présentation clinique ainsi que la prise en charge thérapeutique de la PKHAD sont similaires à la PKRAD.
Conclusion
Les voies biliaires forment un réseau structuré et hiérarchisé de canaux permettant d’assurer la fonction de drainage de la bile du foie vers l’intestin. Son organisation est étroitement contrôlée, dès les phases précoces de l’embryogénèse. En cas d’anomalie de structuration des canaux biliaires au cours du développement embryonnaire, les conséquences peuvent être multiples. Certaines anomalies aboutissent à des variantes anatomiques des voies biliaires, dont l’expression clinique est souvent nulle mais pouvant présenter un risque de plaie biliaire en cas d’intervention chirurgicale. À l’opposé du spectre, d’autres anomalies embryologiques entraînent des conséquences plus sévères, avec un risque de complication biliaire et de cholangiocarcinome important. L’orientation de ces patients vers des centres de référence, composés d’équipes multidisciplinaires expertes, est essentielle pour prévenir la survenue de ces complications graves.
Remerciements :
- Relecture :
- Prs Arrivé
- et O. Chazouillères
- Iconographie :
- Dr Andreani,
- Prs L. Arrivé,
- M. Camus
- et D. Wendum
Références
- Lemaigre Development of the Intrahepatic and Extrahepatic Biliary Tract: A Framework for Understanding Congenital Diseases. Annu Rev Pathol. 2020;15:1-22.
- Kasi PM, Ramirez R, Rogal SS, Littleton K, Fasanella KE. Gallbladder Case Rep Gastroenterol. 2011;5(3):654-62.
- Ye YQ, Liang Q, Li EZ, Gong JL, Fan JM, Wang 3D reconstruction of a gallbladder duplication to guide LC: A case report and literature review. Medicine (Baltimore). 2023;102(8):e33054.
- Chaib E, Kanas AF, Galvão FHF, D’Albuquerque Bile duct confluence: anatomic variations and its classification. Surg Radiol Anat SRA. 2014;36(2):105-9.
- Echchikhi M, Edderai M, Saouab R, Ennouali H, Boumedine H, Radouane B, et Mise au point sur les variantes anatomiques des voies biliaires extra-hépatiques en cholangio-pancréato-IRM et leurs risques de complications. J Imag Diagn Interv. 2021;4(5):307-16.
- Soares KC, Kim Y, Spolverato G, Maithel S, Bauer TW, Marques H, et Presentation and Clinical Outcomes of Choledochal Cysts in Children and Adults: A Multi-institutional Analysis. JAMA Surg. 2015;150(6):577-84.
- Kamisawa T, Ando H, Suyama M, Shimada M, Morine Y, Shimada H, et Japanese clinical practice guidelines for pancreaticobiliary maljunction. J Gastroenterol. 2012;47(7):731-59.
- Brown ZJ, Baghdadi A, Kamel I, Labiner HE, Hewitt DB, Pawlik Diagnosis and management of choledochal cysts. HPB. 2023;25(1):14-25.
- Cazares J, Koga H, Yamataka Choledochal cyst. Pediatr Surg Int. 2023;39(1):209.
- Wu DQ, Zheng LX, Wang QS, Tan WH, Hu SJ, Li Choledochal cysts in pregnancy: case management and literature review. World J Gastroenterol. 2004;10(20):3065-9.
- ten Hove A, de Meijer VE, Hulscher JBF, de Kleine Meta-analysis of risk of developing malignancy in congenital choledochal malformation. Br J Surg. 2018;105(5):482-90.
- Sastry AV, Abbadessa B, Wayne MG, Steele JG, Cooperman What is the incidence of biliary carcinoma in choledochal cysts, when do they develop, and how should it affect management? World J Surg. 2015;39(2):487-92.
- https://afef.asso.fr/recommandation/recommandations-afef-sur-prise-en-charge-des-cholangiocarcinomes-intrahepatiques-et-perihilaires/.
- Jabłońska Biliary cysts: Etiology, diagnosis and management. World J Gastroenterol WJG. 2012;18(35):4801-10.
- Urushihara N, Fukumoto K, Fukuzawa H, Mitsunaga M, Watanabe K, Aoba T, et Long-term outcomes after excision of choledochal cysts in a single institution: operative procedures and late complications. J Pediatr Surg. 2012;47(12):2169-74.
- Ohashi T, Wakai T, Kubota M, Matsuda Y, Arai Y, Ohyama T, et Risk of subsequent biliary malignancy in patients undergoing cyst excision for congenital choledochal cysts. J Gastroenterol Hepatol. 2013;28(2):243-7.
- Mirza H, Besse W, Somlo S, Weinreb J, Kenney B, Jain An update on ductal plate malformations and fibropolycystic diseases of the liver. Hum Pathol. 2023;132:102-13.
- Cnossen WR, Drenth Polycystic liver disease: an overview of pathogenesis, clinical manifestations and management. Orphanet J Rare Dis. 2014;9:69.
- Orphanet: Maladie de Caroli [Internet]. Disponible sur: https://www.orpha.net/fr/disease/detail/53035
- Dhumeaux Lésions kystiques des voies biliaires intra-hépatiques par malformation de la plaque ductale. In: Maladie des voies biliaires. Doin. 2009. (Progrès en hépato-gastroentérologie).
- Diamond T, Nema N, Wen Hepatic Ciliopathy Syndromes. Clin Liver Dis. 2021;18(4):193-7.
- Fahrner R, Dennler SG, Inderbitzin Risk of malignancy in Caroli disease and syndrome: A systematic review. World J Gastroenterol. 2020;26(31):4718-28.
- Gunay-Aygun M, Font-Montgomery E, Lukose L, Tuchman Gerstein M, Piwnica-Worms K, Choyke P, et al. Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney Gastroenterology. 2013;144(1):112-121.e2.
- Ros E, Navarro S, Bru C, Gilabert R, Bianchi L, Bruguera Ursodeoxycholic acid treatment of primary hepatolithiasis in Caroli’s syndrome. Lancet. 1993;342(8868):404-6.
- Wen JW, Furth SL, Ruebner Kidney and liver transplantation in children with fibrocystic liver–kidney disease: Data from the US Scientific Registry of Transplant Recipients: 1990–2010. Pediatr Transplant. 2014;18(7):726-32.
- Gunay-Aygun M, Turkbey BI, Bryant J, Daryanani KT, Gerstein MT, Piwnica-Worms K, et Hepatorenal findings in obligate heterozygotes for autosomal recessive polycystic kidney disease. Mol Genet Metab. 2011;104(4):677-81.
- Su S, Trinh A, Metz AJ, Speer T, Simkin P, Buchanan D, et Targeted liver ultrasound performed by an expert is the pivotal imaging examination for low phospholipid-associated cholelithiasis. Eur J Gastroenterol Hepatol. 2023;35(3):327-32.
- European Association for the Study of the EASL Clinical Practice Guidelines on the management of cystic liver diseases. J Hepatol. 2022;77(4):1083-108.
- Orphanet: Polykystose rénale autosomique dominante [Internet]. Disponible sur: https://www.orpha.net/fr/disease/detail/730
- Masyuk TV, Masyuk AI, LaRusso Polycystic liver disease: The interplay of genes causative for hepatic and renal cystogenesis. Hepatology. 2018;67(6):2462-4.
- Boukoucha M, Dhieb F, Khelifa RB, Znaidi H, Elaifi R, Daghfous Cholecystitis on gallbladder duplication: A case report and literature review. Int J Surg Case Rep. 2020;72:406-10.
- Lobo ML, Stafrace S, Riccabona Imaging the Neonatal GI Tract, Also Including Liver, Spleen, Pancreas, Retroperitoneum and Mesentery. In: Imaging in Neonates. Springer International Publishing; 2023. p. 327-82.
- Morine Y, Shimada M, Ishibashi Most Recent Analysis of a Japanese Nationwide Survey of Pancreaticobiliary Maljunction over a Quarter of a Century. In: Pancreaticobiliary Maljunction and Congenital Biliary Dilatation. Springer; 2018. p. 19-31.