LIENS D’INTÉRÊTS
Abbvie, Gilead, Chiesi, Mayoli, Ipsen, Pfizer, Roche
MOTS-CLÉS
Cirrhose ; Métabolisme hépatique ; Hépatotoxicité
ABRÉVIATIONS
AVK : Anti-Vitamines K
AOD : Anticoagulants oraux directs
CYP : Cytochrome P450
HBPM : Héparines de bas poids moléculaire
INR : International Normalized Ratio (rapport international normalisé)
NATs : N-acétyltransférases
P-gp : P-glycoprotéine SULTs : Sulfotransférases
TFPI : Inhibiteur de la voie du facteur tissulaire
UGT : UDP-glucuronosyltransférases
Introduction
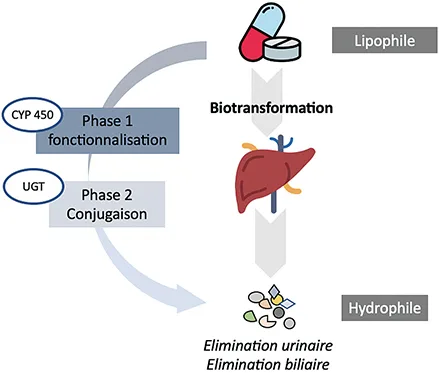
Le métabolisme des médicaments est un processus biologique essentiel qui détermine comment un médicament est transformé dans l’organisme avant d’être éliminé. Il se déroule principalement dans le foie, où les enzymes du cytochrome P450 modifient la structure chimique des médicaments pour les rendre plus solubles et faciliter leur excrétion. Ce métabolisme se déroule généralement en deux phases. La phase I, aussi appelée phase de fonctionnalisation, implique des réactions d’oxydation, de réduction ou d’hydrolyse, qui ajoutent ou exposent des groupes fonctionnels sur la molécule du médicament. La phase II, ou phase de conjugaison, consiste en l’ajout de groupements hydrophiles (comme le glucuronide ou le sulfate) pour rendre le médicament encore plus facilement excrétable par les reins ou la bile (figure 1).
Figure 1 : Métabolisme des médicaments

CYP : cytochrome ; UGT : UDP-glucuronosyltranférases
Tableau 1 : Liste des substrats, inhibiteurs et inducteurs des enzymes impliquées dans le métabolisme médicamenteux (1)
| Enzymes | Substrats | Inhibiteurs (augmente la concentration plasmatique du substrat) | Inducteurs (diminue la concentration plasmatique du substrat) |
| CYP3A4 | Abémaciclib, alectinib, alfuzosine, amiodarone, amlodipine, anastrozole, apixaban, axitinib, budesonide, cabozantinib, carbamazépine, ciclosporine, clarithromycine, clindamycine clonazépam, clopidogrel, colchicine, cyclofosfamide, docetaxel, erythromycine, contraceptifs oraux, everolimus, fentanyl, irinotécan, ivacaftor, ivermectine, ketoconazole, lenvatinib, letrozole, methadone, oxycodone, paclitaxel, palbociclib, pazopanib, prednisone, regorafenib, ribociclib, rifaximine, rivaroxaban, sirolimus, tacrolimus, tamoxifène, upadacitinib, voriconazole, zopicline, zolpidem | Kétoconazole, clarithromycine, erythromycine, fluconazole, itraconazole, posaconazole, jus de pamplemousse, tipranavir/ ritonavir, voriconazole | Carbamazépine, phénobarbital, Phénytoïne, rifampicine, millepertuis |
| CYP1A2 | Caféine, clozapine, dacarbazine, duloxétine, mélatonine, ondansétron, rameltéon, tacrine, tizanidine, warfarine | Aciclovir, ciprofloxacine, fluvoxamine, contraceptifs oraux, phénylpropanolamine, ticlopidine, vemurafenib | Acalabrutinib, binimétinib, carbamazépine, phénobarbital, phénytoïne, rifampicine, tériflunomide, composants du tabac (fumée) |
| CYP2B6 | Cyclophosphamide, ifosfamide, methadone, propofol, tramadol | clopidogrel, ticlopidine, voriconazole | carbamazépine, rifampicine, ritonavir |
| CYP2C8 | Amiodarone, carbamazépine, ibuprofène, imatinib, Répaglinide, paclitaxel, tucatinib, zopiclone | clopidogrel, co-trimoxazole, teriflunomide, triméthoprime | Rifampicine |
| CYP2C19 | Acide valproïque, citalopram, clopidogrel, diazépam, escitalopram, esomeprazole, lanzoprazole, léflunomide, oméprazole, pantoprazole, sertraline, terbinafine, voriconazole, | Fluconazole, fluoxétine, isoniazide, oméprazole, topiramate, voriconazole | efavirenz, rifampicine |
| CYP2D6 | Amitriptyline, aripiprazole, carvédilol, clozapine, codéine, duloxetine, flécaïne, fluoxetine, haloperidol, metoclopramide, nabivolol, oxycodone, propranolol, tamoxifène, tamsulosine, tramadol, venlafaxine | Amiodarone, bupropione, citalopram, duloxétine, excitalopram, fluoxétine, paroxétine, ritanovir, sertraline, terbinafne,venlafaxine | |
| CYP2C9 | Acenocoumarol, acide valproïque, candésartan, carvédilol, Célécoxib, diclofénac, glimepride, gliclazide, ibuprofène, phenobarbital, phénytoïne, rosuvastatine, valsartan, warfarine | Amiodarone, fluconazole, miconazole,capécitabine, fluoxétine, fluvastatine, ivacaftor, voriconazole | Carbamazépine, rifampicine, aprépitant, bosentan, phénobarbital, phénytoïne, millepertuis, rifampicine, ritonavir |
| UDP-glucuronosyl transferases (UGTs) | Bilirubine, phénols, estradiols, opiacés et acides carboxyliques | Paclitaxel, midazolam, cyclosporine A, kétoconazole, phénobarbital, phénytoïne | Bilirubine, phénobarbital, rifampicine |
| sulfotransferases (SULTs) | Phénols, alcools, amines | Flavonoïdes, acides salicyliques, clomiphène, danazol | Acide rétinoïque, méthotrexate |
| N-acetyltransferases (NATs) | Acide para-aminobenzoïque, acides para- aminosalicyliques, paraminoglutamate, sulfaméthazine, isoniazide, hydralazine, sulfamides | Acide caféique, esculétine, quercétine, gentine, scopoline, coumarine | Androgènes, aminophylline |
| Glutathione S-transferases (GSTs) | Époxydes, quinones, sulfoxydes, esters, peroxydes | Phénols, quinone, dérivés de la vitamine C, dopamine, acide rétinoïque | Extraits de brocoli, chou, choux de Bruxelles, pamplemousse |
| P-glycoprotein (P-gp) | Apixaban, atorvastatine, azithromycine, budesonide, carvedilol, cetirizine, ciclosporine, citalopram, clopidogrel, colchicine, dabigatran, digoxine, erytromycine, everolimus, fentanyl, irinotécan,itraconazole, lanvatinib, morphine, ondansétron, odevixibat, paclitaxel, paroxetine, posaconazole, prednisone, rifampicine, rifaximine, rivaroxaban, sertraline, sirolimus, sofosbuvir, ticagrelor, tenofvir, venlaflaxine, voxilaprévir | Amiodarone, azithromycine, cyclosporine, clarithromycine, ketoconazole, érythromycine, itraconazole, kétoconazole, lopinavir/ritonavir, vérapamil | Apalutamide, carbamazépine, ivosidenib, lorlatinib, rifampicine, millepertuis |
L’extraction d’un médicament est un concept pharmacocinétique clé qui reflète la proportion du médicament retirée de la circulation sanguine par un organe, généralement le foie ou les reins, lors de son passage. L’indice d’extraction hépatique (EEE) mesure l’efficacité du foie à extraire un médicament et se calcule comme suit : E=(Centrée−Csortie)/ Centrée où Centrée est la concentration du médicament dans le sang arrivant au foie et Csortie celle quittant le foie. Cet indice varie entre 0 (faible extraction) et 1 (extraction élevée). Les médicaments à forte extraction (EEE proche de 1) sont principalement métabolisés lors d’un premier passage hépatique et leur clairance dépend du débit sanguin hépatique, tandis que les médicaments à faible extraction (EEE proche de 0) sont influencés par l’activité enzymatique et la liaison aux protéines plasmatiques. L’indice d’extraction est déterminant pour estimer la biodisponibilité des médicaments administrés par voie orale, car un effet de premier passage hépatique important peut réduire la quantité disponible pour la circulation systémique.
Le foie en raison de sa vascularisation et de sa richesse en enzymes joue un rôle primordial par rapport aux autres organes : c’est le principal site de métabolisme des médicaments. Environ 70-80 % des médicaments sont métabolisés par le foie. Au niveau cellulaire, l’essentiel de l’activité de métabolisation des médicaments se déroule dans le réticulum endoplasmique (R.E) et dans le cytosol, bien que les biotransformations puissent également se produire à moindre degré dans les mitochondries, la membrane nucléaire et la membrane cytoplasmique.
De multiples facteurs peuvent influencer le métabolisme hépatique des médicaments : caractéristiques physiologiques de l’individu (âge, sexe, grossesse, diabète…), certains facteurs génétiques, présence d’états pathologiques comme la cirrhose ou encore interactions médicamenteuses. Les interactions médicamenteuses peuvent entrainer :
Une induction enzymatique : administration d’une substance étrangère à l’organisme suivie d’une synthèse accrue de l’enzyme qui la dégrade. La substance est alors catabolisée plus rapidement. Les conséquences cliniques de l’induction du métabolisme d’un médicament peuvent être variables :
- Si la formation de métabolites inactifs est accélérée, la durée de l’effet thérapeutique sera raccourcie.
- Si l’induction conduit à une augmentation de métabolites actifs, l’effet thérapeutique sera accru.
- Si la formation de métabolites toxiques est augmentée, des effets indésirables graves peuvent survenir.
Exemples d’inducteurs enzymatique : Phénobarbital ; Carbamazépine ; Rifampicine ; Millepertuis.
Une inhibition enzymatique : c’est la diminution de l’activité des enzymes de métabolisation causée par des substances exogènes qu’elles soient médicamenteuses ou non. Les conséquences cliniques de l’inhibition du métabolisme des médicaments sont :
- Une augmentation de la concentration de produit actif et une augmentation de la durée de l’effet thérapeutique avec un risque de toxicité si la formation de métabolites inactifs est ralentie par l’inhibition enzymatique. C’est le cas le plus fréquent.
- Une diminution de l’effet thérapeutique si l’inhibition conduit à une diminution de la formation de métabolites actifs.
Exemples d’inhibiteurs enzymatiques : Macrolides ; Cimétidine ; Jus de pamplemousse (contient des flavonoïdes qui sont des inhibiteurs de la CYP3A4).
Chacun des médicaments peut être le substrat d’un isoforme des CYP et également inhibiteur ou inducteur d’un ou plusieurs CYP (tableau 1).
Métabolisme des médicaments et cirrhose (2, 3)
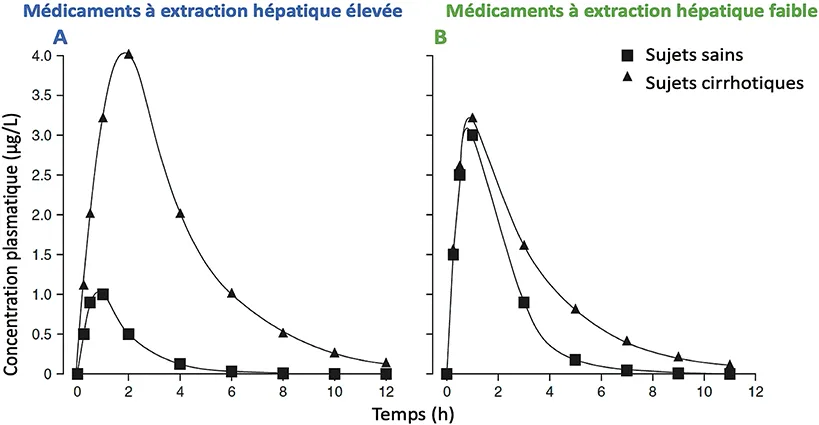
Le foie est le principal organe impliqué dans le métabolisme médicamenteux, en cas de cirrhose des changements vont être observés (figure 2).
Figure 2 : Effet de la cirrhose du foie sur la cinétique des médicaments ayant une extraction hépatique élevée ou faible

A. Médicaments à extraction hépatique élevée : Sujet sain (carrés noirs) : Les concentrations plasmatiques atteignent un pic plus faible, et le médicament est éliminé Sujet avec cirrhose du foie (triangles noirs) : La concentration plasmatique maximale est plus élevée, la biodisponibilité est augmentée, et l’élimination est ralentie.
B. Médicaments à extraction hépatique faible : Sujet sain (carrés noirs) : La concentration maximale est atteinte rapidement, suivie d’une élimination régulière. Sujet avec cirrhose du foie (triangles noirs) : La concentration maximale est légèrement plus élevée, et l’élimination est plus lente.
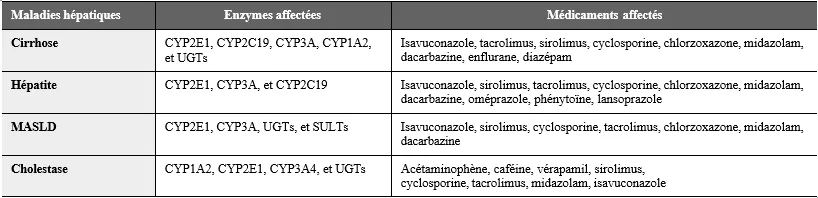
Tableau 2 : Enzymes et métabolisme médicamenteux affectés en fonction des pathologieshépatiques (8)

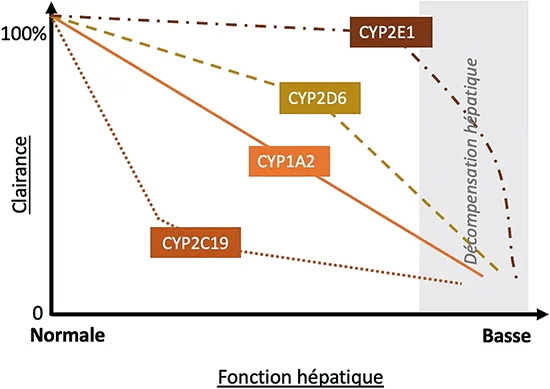
En effet, la cirrhose entraine des modifications du flux sanguin hépatique, une expression altérée des enzymes du métabolisme médicamenteux, une disponibilité modifiée des co-substrats et une liaison altérée des médicaments aux protéines plasmatiques (1). Les enzymes oxydatives du cytochrome P450 (CYP450) nécessitent de l’oxygène pour fonctionner, et celui-ci est moins disponible cas de shunt porto-systémique ou de perfusion hépatique diminuée. En général, le métabolisme diminue parallèlement à la détérioration de la fonction hépatique (tableau 2). Cependant, la corrélation entre quantification du métabolisme hépatique et les scores d’évaluation de la fonction hépatique (MELD, CHILD-PUGH) n’est pas évidente (4). L’activité de certaines isoenzymes du CYP450 peut être relativement préservée lorsque la fonction hépatique diminue, comme le CYP2E1, tandis que celle d’autres isoenzymes, comme le CYP2C19, est relativement sensible aux dysfonctionnements hépatiques (5, 6) (figure 3). Le shunt porto-systémique, qu’il soit endogène ou iatrogène (TIPS), peut augmenter la biodisponibilité orale en raison de la réduction du métabolisme de premier passage dans la cirrhose. Le métabolisme par glucuronidation est moins affecté que les isoenzymes du CYP450, peut- être en raison de la glucuronidation extra-hépatique ou de l’augmentation de l’expression de l’uridine diphosphate-glucuronyltransférase dans les hépatocytes viables restants. Enfin, l’excrétion biliaire des médicaments et de leurs métabolites est diminuée en cas d’obstruction biliaire, de malignité et de maladies hépatiques cholestatiques, et les lésions hépatocellulaires qui surviennent avec l’obstruction biliaire peuvent réduire l’activité des isoenzymes du CYP450.
Figure 3 : Baisse de la clairance des CYP en fonction de la fonction hépatique (9)

Pour chaque médicament, le Résumé des Caractéristiques du Produit (RCP) précise les précautions à prendre en cas de prescription chez un patient cirrhotique. La principale limite de ces données est l’exclusion des patients cirrhotiques des essais thérapeutiques et le peu de données pharmacologiques dans ces états pathologiques. En cas de prescription chez le patient cirrhotique, les médicaments pour lesquels une attention particulière doit être portée, sont ceux ayant une faible extraction hépatique et une marge thérapeutique étroite. Si ces médicaments sont administrés par voie orale, les doses initiales et d’entretien doivent être réduites d’au moins 50 % de la dose normale, en fonction de la gravité de la maladie hépatique, de l’extraction et du métabolisme hépatique, ainsi que de la toxicité du médicament (7).
Hépatotoxicité des médicaments et cirrhose (10)
L’hépatotoxicité médicamenteuse est une complication qui concerne une grande variété de produits de santé, avec des mécanismes, des fréquences et des degrés de sévérité variables. Chez les patients atteints de cirrhose, l’altération du métabolisme hépatique et des mécanismes de clairance peut potentialiser la toxicité de certains médicaments, comme cela a été documenté pour le paracétamol, bien que cette susceptibilité doive être évaluée au cas par cas en fonction des données pharmacocinétiques et de toxicité. Par ailleurs, la survenue d’une hépatite aiguë, indépendamment de son étiologie, peut induire une décompensation hépatique chez des patients présentant une cirrhose sous-jacente, avec des conséquences cliniques potentiellement graves. L’identification des risques liés à une substance donnée est facilitée par l’utilisation de bases de données spécialisées comme LiverTox ou par la consultation des services de pharmacovigilance. Ces considérations soulignent la nécessité d’une approche thérapeutique prudente et individualisée chez les patients cirrhotiques afin de minimiser le risque de toxicité médicamenteuse et d’événements indésirables.
Les risques et les précautions d’usage des médicaments antalgiques en cas de cirrhose
La gestion de la douleur chez les patients cirrhotiques est une problématique récurrente. Afin de fournir une analgésie efficace et de réduire le risque de complications, il est important de prendre en compte l’impact de la cirrhose sur le métabolisme et l’action des agents antalgiques.
Paracétamol
Le paracétamol, antalgique de palier I disponible sans ordonnance, est connu pour son hépatotoxicité en cas de surdosage. Il est métabolisé par le foie via la glucuronidation et la sulfatation en métabolites non toxiques, un processus qui reste bien conservé en cas de cirrhose. Seule une petite proportion (< 10 %) est métabolisée par le CYP2E1, produisant l’intermédiaire hépatotoxique N-acétyl-p-benzoquinone imine, qui est ensuite conjugué avec le glutathion (11). Une hépatite peut survenir en cas de surdosage de paracétamol et, dans de rares cas, suite à des doses thérapeutiques répétées chez des patients présentant des facteurs de risque de carence en glutathion, comme la malnutrition, les maladies chroniques ou une consommation chronique d’alcool. Il n’y a aucune preuve que le paracétamol contribue à la décompensation hépatique (12). De même, les études de pharmacocinétique du paracétamol chez les patients cirrhotiques comportent de nombreux biais, mais ne sont pas en faveur d’un risque accru (13). Les recommandations ne contre indiquent pas l’usage du paracétamol chez les patients cirrhotiques mais proposent de limiter la posologie à 2-3 g par jour, et ce d’autant plus que le patient a une consommation chronique d’alcool ou une insuffisance rénale associée.
Le néfopam
Le néfopam est un analgésique non opioïde principalement métabolisé par le foie via les enzymes du cytochrome P450, notamment le CYP2C19 et le CYP1A2, pour être excrété sous forme de métabolites inactifs. Chez les patients atteints de cirrhose, la diminution de la clairance hépatique peut entraîner une accumulation du néfopam, augmentant le risque d’effets indésirables graves tels que confusion, hallucinations, convulsions ou hypertonie. Bien que les données sur l’utilisation du néfopam chez les patients cirrhotiques soient limitées, il est considéré comme le moins risqué parmi les antalgiques des paliers 1 et 2.
Les AINS
Les anti-inflammatoires non stéroïdiens (AINS) sont majoritairement métabolisés par les enzymes du cytochrome P450 (CYP) et présentent une forte liaison aux protéines plasmatiques. Ainsi, chez les patients cirrhotiques, on peut s’attendre à un métabolisme altéré et à une biodisponibilité accrue, entraînant une élévation des concentrations sériques. La principale préoccupation lors de l’usage des AINS chez le cirrhotique est le risque de provoquer ou d’aggraver une dysfonction rénale. Les AINS sont également des causes fréquentes de saignements gastro-intestinaux et ont été associés au premier épisode de saignement variqueux chez les patients atteints de cirrhose et d’hypertension portale dans une étude rétrospective cas-témoins, possiblement en raison de lésions muqueuses chez des patients à risque accru de saignement (14). Enfin, les AINS sont une cause fréquente d’hépatototoxicité (10). La toxicité est idiosyncrasique, elle n’est pas dose dépendante, elle peut être sévère et entraîner une décompensation de cirrhose. Les AINS peuvent être tolérés chez les patients atteints d’une maladie hépatique chronique non sévère, mais ils doivent être évités chez tous les patients atteints de cirrhose (11).
Les opiacés (11, 15, 16)
Les opiacés doivent être utilisés avec prudence chez les patients atteints de cirrhose, car ils peuvent entraîner de l’encéphalopathie hépatique. Ils sont principalement métabolisés via le CYP450 (CYP2D6 et CYP3A4) et par glucuronidation, deux processus affectés en cas de cirrhose. Le métabolisme de la morphine est considérablement altéré dans les maladies hépatiques sévères, avec une réduction de la clairance plasmatique, une demi-vie prolongée et une augmentation de la biodisponibilité orale par rapport aux patients ayant une fonction hépatique normale. De même, l’hydromorphone présente une biodisponibilité accrue en cas de maladie hépatique. Les opioïdes endogènes peuvent être régulés à la hausse en cas de cholestase, accentuant les effets sédatifs des opioïdes exogènes.
L’utilisation de la codéine et de l’oxycodone chez les patients atteints de maladie hépatique peut entraîner une analgésie variable, car le métabolite actif n’est pas toujours formé de manière fiable, et une élimination lente peut provoquer une accumulation du médicament et une dépression respiratoire. Les formules à libération prolongée doivent être évitées chez les patients atteints de maladie hépatique, car la demi-vie est déjà augmentée.
La buprénorphine est un agoniste partiel des récepteurs mu-opioïdes, principalement métabolisé par le foie via le CYP450, y compris le CYP3A4, en métabolite actif norbuprénorphine, puis éliminé par glucuronidation et excrété dans la bile. Le fentanyl peut être mieux toléré par les patients cirrhotiques, car sa demi-vie d’élimination n’est pas modifiée. La méthadone est souvent utilisée chez les patients atteints de maladie hépatique chronique avec une dépendance aux opioïdes. Les indices pharmacocinétiques de la méthadone chez les patients présentant une maladie hépatique stable sont similaires à ceux des témoins sains, ce qui suggère que la posologie de maintien ne doit pas être modifiée, bien que chez les patients atteints de maladie hépatique sévère, la demi-vie soit plus longue. Le fentanyl et la méthadone sont fortement liés aux protéines et nécessitent une réduction de la dose chez les patients souffrant d’hypoalbuminémie.
Tramadol
Le tramadol est un opioïde parfois utilisé à faibles doses chez les patients atteints de cirrhose souffrant de douleurs réfractaires. Son mécanisme d’action repose sur son impact sur les voies de la douleur périphérique, son inhibition partielle de la recapture de la sérotonine et sa faible affinité pour les récepteurs opioïdes. Ces caractéristiques sont supposées réduire les effets de sédation, la dépression respiratoire et le risque de tolérance.
L’insuffisance rénale due au syndrome hépatorénal complique le métabolisme des médicaments, en particulier pour des médicaments tels que le paracétamol, la codéine, l’oxycodone et la morphine, qui sont principalement éliminés par les reins.
Tableau 3 : Métabolisme et excrétion des antalgiques (11, 15, 16)
| Médicament | Demi-vie (h) | Liaison protéique (%) | Métabolisme hépatique | Excretion |
| Tramadol | 5.1 (sujet sain), 13.3 (cirrhose) | 20 | CYP3A4, CYP2D6, glucuronidation | rénale (60%) |
| Hydrocodone | 3.8 | 7 | CYP2D6 | rénale (26%) |
| Oxycodone | 3.4 (sujet sain), 13.9 (cirrhose) | 45 | CYP3A4, 2D6 | rénale |
| Morphine | 3.3 (sujet sain), 5.5 (cirrhose) | 35 | Glucuronidation | rénale (90%) / fécale (7-10%) |
| Hydromorphone | 2.5 | 15 | Glucuronidation | rénale (1-13% inchangée et 22-51% conjuguée) |
| Méthadone | 19 (sujet sain), 35 (cirrhose) | 80 | CYP3A4, 2D6, et plusieurs autres | rénale (21%) / fécale / biliaire |
| Codéine | 2.9 (sujet sain) | 7 | CYP2D6, glucuronidation | rénale (90%) / fécale (10%) |
| Mépridine | 2-4 | 70 | CYP2B6, 3A4 | rénale |
| Fentanyl IV | 3-4 | 7 | CYP3A4 | rénale (75%) / fécale |
| Fantanyl Patch | 17 | 80 | CYP3A4 | rénale (75%) / fécale |
| Bupremorphine | | 96 | CYP3A4, glucuronidation | biliaire |
| Paracetamol | 1,5-3 | 25 | CYP2E1 (10%), glucuronidation (60%), sulfoconjugaison (30%) | rénale (90%) |
| Nefopam | 4 | 71-76% | CYP2C19, CYP1A2 et le CYP2D6 | rénale (87%) |
La pharmacodynamie des analgésiques peut également être modifiée en cas de cirrhose, avec des effets accrus sur le système nerveux central des opiacés, sédatifs et anxiolytiques, pouvant précipiter une encéphalopathie. L’étiologie précise de ce phénomène est inconnue, mais il est supposé qu’elle soit liée à des altérations de la perméabilité de la barrière hémato-encéphalique, à une neurotransmission médiée par l’acide gamma- aminobutyrique (GABA) augmentée et/ou à une densité, une affinité ou une sensibilité accrue des récepteurs aux médicaments.
Gestion des différentes classes d’anticoagulants et anti agrégants en cas de cirrhose
Les anticoagulants (17)
Pendant de nombreuses années, les patients atteints de cirrhose ont été considérés comme « auto-anticoagulés », une hypothèse basée sur une interprétation erronée de l’allongement des temps de coagulation (notamment le temps de prothrombine et le temps de thromboplastine partielle activée). Ces paramètres, bien que sensibles à une diminution des facteurs de coagulation, ne reflètent pas une réduction des anticoagulants naturels. Cependant, d’un point de vue hémostatique, des données de plus en plus nombreuses au cours de la dernière décennie suggèrent un rééquilibrage de l’hémostase chez les patients cirrhotiques. Par ailleurs, des études cliniques ont révélé une augmentation des événements thromboemboliques veineux, en particulier des thromboses splanchniques et des thromboses de la veine porte, dont le risque est significativement accru aux stades avancés de la cirrhose.
AVK
Les anti-vitamines K (AVK) agissent en inhibant les facteurs de coagulation dépendants de la vitamine K (II, VII, IX, X) ainsi que les anticoagulants naturels, tels que la protéine C et la protéine S. Chez les patients atteints de cirrhose, le rapport international normalisé (INR) est souvent déjà élevé au départ, ce qui reflète une diminution de la synthèse hépatique des facteurs de coagulation, y compris le facteur V, qui est indépendant de la vitamine K. Cette altération rend le suivi de l’efficacité et de la sécurité des AVK particulièrement complexe dans ce contexte.
Héparines de bas poids moléculaire (HBPM)
Une diminution du potentiel anticoagulant des HBPM peut être anticipée, car leur activité biologique repose sur l’antithrombine, dont les niveaux sont souvent réduits dans un contexte de synthèse hépatique protéique compromise. Cependant, cette hypothèse a été nuancée par les travaux de Senzolo et al., qui ont rapporté une réponse anticoagulante paradoxalement accrue aux HBPM chez les patients atteints de cirrhose. Ces résultats, ainsi que d’autres études, ont mis en évidence un risque de surdosage dans cette population, en raison d’une activité anti-Xa souvent inférieure à celle attendue pour le rapport dose/poids corporel. Le potentiel anti-thrombotique augmenté des HBPM en cas de cirrhose pourrait être attribué à l’action anticoagulante indépendante de l’antithrombine exercée par l’inhibiteur de la voie du facteur tissulaire (TFPI). Ce dernier, un anticoagulant naturel majoritairement stocké dans l’endothélium, inhibe le facteur VII activé lié au facteur tissulaire. Concernant le risque de surdosage chez les patients atteints de cirrhose décompensée, il est essentiel de prendre en compte la surestimation du poids corporel due à l’accumulation de fluides (par exemple, ascite) ainsi que l’altération de l’élimination rénale des HBPM, souvent observée dans des situations telles que l’insuffisance rénale aiguë ou le syndrome hépatorénal. Malgré ces considérations, les HBPM demeurent un traitement de référence chez les patients cirrhotiques, grâce à leur efficacité, leur sécurité d’utilisation et leur profil bien documenté. De plus, la surveillance rapprochée de l’activité anti-Xa est rarement nécessaire dans ce contexte.
Anticoagulants oraux direct (AOD)
L’absorption systémique des AOD chez les patients atteints de cirrhose semble globalement préservée, en raison d’un effet de premier passage insignifiant. Néanmoins, des concentrations plasmatiques plus élevées d’AOD peuvent être attendues dans cette population pour plusieurs raisons. La dysfonction hépatique s’accompagne souvent de niveaux réduits d’albumine, entraînant une augmentation de la fraction active non liée des AOD fortement liés aux protéines plasmatiques, comme l’apixaban et le rivaroxaban. Une diminution de la clairance des AOD est également attendue, en particulier pour ceux métabolisés par les cytochromes P450 (CYP450), notamment les inhibiteurs directs du FXa (rivaroxaban, apixaban et édoxaban). Ces inhibiteurs sont principalement métabolisés par le CYP3A4, et dans une moindre mesure par d’autres enzymes du système CYP450. Cependant, l’impact du système CYP450 reste difficile à prédire, car la sensibilité de chaque cytochrome est spécifique à chaque individu et peut être davantage altérée selon le stade de la cirrhose. Cette variabilité individuelle et l’évolution de la fonction hépatique selon la progression de la cirrhose nécessitent une attention particulière lors de l’utilisation des AOD chez ces patients.
Tableau 4 : Métabolisme et recommandations d’utilisation des AOD
| Paramètres | Rivaroxaban | Apixaban | Edoxaban | Dabigatran |
| Cible | Facteur X activé | Facteur X activé | Facteur X activé | Facteur II activé |
| Pro-médicament | Non | Non | Non | Oui, converti dans le plasma et le foie en sa forme active |
| Biodisponibilité (%) | 66 (sans nourriture) 80-100 (avec nourriture pour RVX 15 et 20 mg) | 50 | 62 | 6-7 |
| Demi-vie (h) | 5-9 | 8-15 | 10-14 | 12-14 |
| Liaison aux protéines (%) | 92 | 87 | 55 | 35 |
| Métabolisme via CYP450 (%) | 66 | 25 | 50 | Aucun |
| Type de CYP450 | CYP3A4, CYP2J2 | CYP3A4/5 (implication majeure), CYP1A2 (mineure), CYP2C8/2C9/2C19/2J2 (mineures) | CYP3A4/5 | Conjugué avec l’acide glucuronique |
| Voies d’élimination (%) | Excrétion hépatobiliaire/ fécale 33% Rénale 66% (inchangée et 30% sous forme de métabolites) | Excrétion hépatobiliaire/ fécale 75% Rénale 25% (majoritairement inchangée) | Excrétion hépatobiliaire/ fécale 50% Rénale 50% (majoritairement inchangée) | Excrétion hépatobiliaire/ fécale 20% Rénale 80% (majoritairement inchangée) |
| Dose thérapeutique (Standard) | 20 mg | 2×5 mg | 60 mg | 2×150 mg |
| Dose thérapeutique (Ajustée/réduite) | 15 mg | 2×2.5 mg | 30 mg | 2×110 mg |
| AUC en cas de cirrhose CHILD A | +15% | +3% | -4.2% | Pas de données |
| AUC en cas de cirrhose CHILD B | +227% | +93% | -4.8% | -6% |
| AUC en cas de cirrhose CHILD C | Éviter | Pas de données | Éviter | Éviter |
| Recommandations pour l’administration du médicament si cirrhose CHILD A | Dose standard | Dose standard | Dose standard | Dose standard |
| Recommandations pour l’administration du médicament si cirrhose CHILD B | Éviter | Utiliser avec prudence | Utiliser avec prudence | Utiliser avec prudence |
| Recommandations pour l’administration du médicament si cirrhose CHILD C | Éviter | Éviter | Éviter | Éviter |
En résumé, sur la seule base des données pharmacocinétiques et pharmacodynamiques limitées publiées concernant les patients atteints de cirrhose, nous ne pouvons formuler que des hypothèses sur l’adéquation et les adaptations posologiques des AOD (anticoagulants oraux directs) dans cette population. Selon ces données, l’apixaban et l’édoxaban semblent être appropriés pour les patients atteints de cirrhose, y compris ceux présentant une cirrhose classée Child B (avec prudence). En revanche, ils sont contre indiqués chez les patients avec cirrhose CHILD C.
Les antiagrégants (18)
L’aspirine, un antiplaquettaire (AP) disponible en vente libre, est utilisée à des fins médicales depuis de nombreuses décennies. Plus récemment, une nouvelle classe de médicaments antiplaquettaires, les inhibiteurs du récepteur P2Y12 – à savoir le clopidogrel, le ticagrelor et le prasugrel – a été introduite chez les patients présentant des comorbidités cardiaques, y compris ceux atteints de cirrhose. Comme pour les anticoagulants, une thrombocytopénie sévère, définie par un taux de plaquettes inférieur à 50 000/mm3, constitue une contre-indication relative à l’utilisation de ces médicaments en raison du risque élevé de saignement.
Les études pharmacocinétiques de l’aspirine chez les patients présentant une insuffisance hépatique sont très limitées. Dans une petite étude sur une dose unique, la pharmacocinétique de l’aspirine a été comparée entre des sujets jeunes et âgés en bonne santé, et des patients atteints de maladie hépatique liée à l’alcool. L’exposition globale au médicament, la concentration plasmatique maximale et la demi-vie de l’aspirine ne différaient pas de manière significative entre les patients atteints de maladie hépatique alcoolique et les participants en bonne santé.
Clopidogrel
Les recommandations de la FDA (Food and Drug Administration) et de l’EMA (European Medicines Agency) diffèrent concernant l’utilisation du clopidogrel en cas d’insuffisance hépatique. Alors que la FDA recommande de ne pas ajuster la posologie chez les patients atteints d’insuffisance hépatique, l’EMA indique que le clopidogrel doit être utilisé avec prudence chez les patients atteints de maladie hépatique modérée et est contre- indiqué en cas d’insuffisance hépatique sévère. Une étude a exploré l’utilisation du clopidogrel chez les patients atteints de cirrhose. Slugg et al. ont mené une étude ouverte avec 12 patients cirrhotiques (Child-Pugh A/B) et 12 témoins non cirrhotiques appariés. Après 10 jours de traitement, les auteurs n’ont observé aucune différence statistiquement significative en termes d’agrégation plaquettaire et de prolongation du temps de saignement. Aucun événement hémorragique n’a été signalé.
Prasugrel
La FDA et l’EMA recommandent toutes deux l’utilisation du prasugrel sans ajustement posologique chez les patients atteints d’insuffisance hépatique légère à modérée. Une étude pharmacocinétique menée par Small et al. ont révélé que l’insuffisance hépatique modérée n’avait pas d’effet significatif sur l’exposition au métabolite actif du prasugrel, avec peu ou pas d’effet sur l’agrégation plaquettaire par rapport aux témoins sains.
Ticagrélor
Pour les patients présentant une insuffisance hépatique légère à modérée, les deux agences recommandent l’utilisation du ticagrélor sans ajustement posologique. Butler et al. ont comparé les effets du ticagrélor chez des patients atteints d’insuffisance hépatique légère à ceux de témoins sains. Bien que l’exposition au ticagrélor ait été plus élevée chez les patients présentant une insuffisance hépatique légère, cela n’a pas influencé l’agrégation plaquettaire ni la tolérance.
Savoir utiliser les différentes classes de traitement anti diabétiques et hypolipémiants
Les antidiabétiques (tableau 5)
L’association diabète et cirrhose est bien connue. Certains antidiabétiques, tels que la metformine et les agonistes des récepteurs GLP-1, présentent des effets bénéfiques pour le foie. Cependant, l’utilisation de ces traitements doit être accompagnée de précautions chez les patients atteints de cirrhose décompensée. En effet, les patients avec une cirrhose Child B ou C, sont plus à risque de présenter des hypoglycémies nocturnes. Par ailleurs, des travaux ont mis en évidence chez les patients diabétiques, une diminution significative de l’activité enzymatique du CYP3A4.
Biguanides (Metformine)
La metformine reste un traitement de première ligne dans de nombreux cas, même chez les patients cirrhotiques, sauf en cas de décompensation avancée (Child-Pugh C) ou d’insuffisance rénale sévère, où elle est contre-indiquée en raison du risque d’acidose lactique. Il est recommandé de ne pas dépasser la dose de 1000 mg par jour chez les patients cirrhotiques et d’arrêter la metformine en cas de décompensation aiguë. Un essai de pharmacocinétique conduit chez les patients cirrhotiques a rapporté des données de sécurité rassurantes chez ces patients, notamment une absence d’augmentation des lactates (19).
Sulfamides hypoglycémiants
Les sulfamides, comme le glibenclamide et le gliclazide, peuvent être utilisés avec prudence, mais ils comportent un risque accru d’hypoglycémie, particulièrement en cas d’insuffisance hépatique. L’hypoglycémie peut être prolongée en raison d’une diminution de la clairance hépatique. Il est donc recommandé de préférer des sulfamides à courte demi-vie (gliclazide ou glipizide) et d’éviter ceux à longue demi-vie (glibenclamide).
Inhibiteurs de la DPP-4 (gliptines)
Les gliptines (sitagliptine, vildagliptine, linagliptine) sont globalement bien tolérées en cas de cirrhose non décompensée (Child-Pugh A ou B), car elles présentent un faible risque d’hypoglycémie. Cependant, certaines nécessitent un ajustement posologique en cas d’insuffisance rénale ou hépatique.
Analogues du GLP-1 (agonistes des récepteurs GLP-1)
Les agonistes du GLP-1 (ex. : liraglutide, dulaglutide, semaglutide) peuvent être utilisés chez les patients cirrhotiques non décompensés (Child- Pugh A ou B). Les données restent limitées pour leur utilisation en cas de cirrhose avancée et une attention particulière doit être portée au risque de dénutrition et de sarcopénie pouvant impacter le pronostic en cas de cirrhose avancée.
Tableau 5 : Anti-diabétiques et cirrhose
| Cirrhose | Child A | Child B | Child C |
| Metformine (si créatininémie normale) | Oui | Non | Non |
| Sulfamide et glinides | Non | Non | Non |
| Analogue GLP1 | Oui | Non | Non |
| iGLT2 | Oui | Non | Non |
| Insuline | Oui | Oui | Oui |
Inhibiteurs des SGLT2 (gliflozines)
Les inhibiteurs des SGLT2 (ex. : empagliflozine, dapagliflozine) présentent des avantages cardiovasculaires et rénaux, mais leur utilisation en cas de cirrhose est limitée. Ils doivent être utilisés avec une grande prudence, car ils peuvent aggraver une déshydratation et un syndrome hépatorénal. Tout comme les analogues GLP1 une attention particulière doit être portée au risque de dénutrition et de sarcopénie pouvant impacter le pronostic en cas de cirrhose avancée.
Insuline
L’insuline reste une option incontournable, notamment chez les patients atteints de cirrhose décompensée ou en cas de diabète mal contrôlé. Cependant, le métabolisme de l’insuline est altéré en cas d’insuffisance hépatique, augmentant le risque d’hypoglycémie car ces patients n’ont aucune réserve de glycogène hépatique. Il est donc préféré d’utiliser des schémas basaux-bolus pour ajuster les doses selon les apports alimentaires, de surveiller fréquemment la glycémie pour prévenir les hypoglycémies et d’utiliser des doses réduites en cas de décompensation hépatique.
Le choix d’un traitement antidiabétique chez les patients cirrhotiques nécessite une approche individualisée tenant compte de la gravité de la cirrhose, des comorbidités et des effets secondaires potentiels. Une surveillance étroite des paramètres hépatiques, rénaux et de la glycémie est essentielle pour optimiser la sécurité et l’efficacité du traitement. En cas de doute, l’insuline reste une option sûre, notamment chez les patients avec une cirrhose avancée ou décompensée.
Tableau 6 : Essais thérapeutiques évaluant l’intérêt des statines chez les patients cirrhotiques
| Étude (auteurs, journal, année) | Nombre de patients | Type/Dosage de statine | Durée d’utilisation des statines | résultats / Commentaires |
| Abrales et al., Gastroenterology, 2016 | 69 ont reçu des statines, 78 ont reçu un placebo | Simvastatine 40 mg | 2 ans | Résultat non significatif pour le critère principal. Risque plus élevé de rhabdomyolyse chez les patients atteints de maladie hépatique avancée. |
| Abrales et al., Gastroenterology, 2009 | 29 ont reçu des statines, 30 ont reçu un placebo | Simvastatine 40 mg | 1 mois | Aucune différence significative dans les événements indésirables ; patients atteints de maladie hépatique avancée exclus. |
| Pollo-Flores et al., Digestive and Liver Disease, 2015 | 14 ont reçu des statines, 20 ont reçu un placebo | Simvastatine 40 mg | 3 mois | Aucun événement indésirable significatif. |
| Elwan et al., F1000 Research, 2018 | 20 ont reçu des statines, 20 ont reçu un placebo | Simvastatine 20 mg pendant 2 semaines ; puis 40 mg pour les 2 semaines restantes | 1 mois | Aucune différence significative dans les événements indésirables. |
| Bishnu et al., European Journal of Gastroenterology and Hepatology, 2018 | 12 ont reçu du propranolol seul, 12 ont reçu de l’atorvastatine et du propranolol | Atorvastatine 20 mg | 1 an | Aucune différence dans les résultats cliniques à 1 an |
| Wani et al., World Journal of Hepatology, 2017 | 62 répondeurs carvédilol, 35 non-répondeurs carvédilol ayant reçu de la simvastatine | Simvastatine 40 mg | 3 mois | Aucune différence significative dans les événements indésirables. |
| Vijayaraghavan et al., American Journal of Gastroenterology, 2020 | 110 carvédilol seul, 110 carvédilol plus simvastatine | Simvastatine 20 mg pendant 2 semaines ; puis 40 mg pour les 2 semaines restantes | 3 mois | Résultat non significatif pour le critère principal. Trois patients sous simvastatine (3.7%) ont développé des transaminases transitoires et une élévation de la créatine kinase. |
Les statines
L’utilisation des statines chez les patients cirrhotiques est souvent questionnée avec d’une part des bénéfices sur la progression de la fibrose, la réduction de la pression portale, la diminution du risque de décompensation hépatique et l’amélioration de la survie ; d’une autre part le risque d’hépatotoxicité (20). En effet, la toxicité hépatique potentielle des statines est une préoccupation fréquente, limitant leur utilisation chez les patients atteints de maladies hépatiques. Bien que des élévations transitoires et asymptomatiques des transaminases soient courantes chez les utilisateurs de statines, les cas de toxicité hépatique significative sont rares (moins de 2 cas par million de patients par an) (21). Ainsi, les patients avec des maladies hépatiques (à l’exception des cirrhoses décompensées) peuvent utiliser les statines sans risque accru de toxicité hépatique comparé à la population générale. Un autre effet secondaire préoccupant est la toxicité musculaire, pouvant atteindre la rhabdomyolyse dans sa forme sévère (22). Les patients atteints de cirrhose avancée sont plus exposés aux effets indésirables, en raison de mécanismes tels que la réduction de l’activité du CYP3A4 (limitant le métabolisme des statines), l’altération des transporteurs MRP2 et SLCO1B, et la réduction de l’absorption hépatique. Dans une étude récente, 3 % des patients avec une cirrhose décompensée avancée prenant 40 mg de simvastatine ont développé une rhabdomyolyse, contre un taux de 0,1 % dans la population générale (23). Une autre étude a montré une toxicité musculaire dose-dépendante avec l’utilisation combinée de simvastatine et rifaximine (24). Aucun des six autres essais thérapeutiques n’a observé d’événements indésirables graves liés aux statines graves liés aux statines ; cependant, ces études excluaient toutes les patients cirrhotiques avec une bilirubine> 5 mg/l, avaient des échantillons de petite taille, et une durée de suivi généralement plus courte (tableau 5). Actuellement, aucune formulation spécifique de statines n’est plus recommandée que les autres chez les patients cirrhotiques. La dose minimale efficace doit être utilisée pour atteindre les objectifs cardiovasculaires, et les statines doivent être arrêtées en cas d’effets secondaires graves.
Les fibrates
Les fibrates ne sont pas contre indiqués en cas de cirrhose compensée, en revanche ils sont plutôt déconseillés en cas de cirrhose décompensée en raison du risque d’hépatotoxicité et de rhabdomyolyse. Ils sont principalement métabolisés par glucurono-conjugaison et éliminés par voie rénale. Alors qu’ils étaient initialement indiqués en raison de leur propriété hypolipémiante, ils sont depuis quelques années recommandés en 2e ligne pour le traitement de la cholangite biliaire primitive (CBP). Récemment une équipe a rapporté, son expérience chez les patients traités par fibrates pour une CBP au stade de cirrhose parfois décompensée. L’effectif de patients est restreint mais la tolérance était bonne (25).
Connaître les règles de bon usage des antibiotiques (1, 26)
Les complications infectieuses sont fréquentes chez le patient cirrhotique, c’est donc une classe médicamenteuse régulièrement prescrite. L’augmentation des germes résistants chez les patients cirrhotiques a pour conséquence parfois l’utilisation de plusieurs lignes d’antibiothérapie avant d’obtenir l’efficacité clinique requise. Enfin il s’agit d’une des classes médicamenteuses la plus reconnue pour son risque hépatotoxique.
Les bêta-lactamines, comprenant les pénicillines, céphalosporines et carbapénèmes, sont principalement éliminées par voie rénale avec une métabolisation hépatique minime, ce qui les rend généralement sûres en cas de cirrhose. Aucun ajustement de dose n’est nécessaire chez ces patients, mais l’utilisation prolongée des céphalosporines de troisième génération, comme la ceftriaxone, doit être évitée chez les patients avec une ascite importante, en raison du risque de précipitation biliaire. Les carbapénèmes, comme l’imipénème, doivent être réservés aux infections sévères résistantes, car ils peuvent perturber le microbiote intestinal.
Les aminosides, tels que la gentamicine et l’amikacine, sont éliminés par voie rénale sans métabolisation hépatique. Leur utilisation est limitée chez les patients cirrhotiques, notamment en cas d’insuffisance rénale ou de syndrome hépatorénal, en raison d’un risque élevé de néphrotoxicité. Une surveillance étroite des taux plasmatiques est nécessaire pour prévenir les toxicités.
Les macrolides, comme l’azithromycine et la clarithromycine, sont métabolisés par le foie via le CYP450, en particulier le CYP3A4, ce qui peut entraîner une accumulation chez les patients atteints de cirrhose avancée. Ils doivent être utilisés avec prudence dans ces cas, et l’azithromycine est préférable en raison de son métabolisme hépatique moindre et de son meilleur profil de sécurité.
Les fluoroquinolones, comprenant la ciprofloxacine et la lévofloxacine, sont partiellement métabolisées par le foie mais majoritairement éliminées par voie rénale. La ciprofloxacine est souvent utilisée pour la prophylaxie et le traitement de la péritonite bactérienne spontanée (PBS) chez les patients avec ascite. Cependant, une réduction de dose est nécessaire en cas d’insuffisance rénale associée, et une utilisation prolongée doit être évitée pour limiter le risque de résistance bactérienne.
Les tétracyclines, comme la doxycycline, sont métabolisées par le foie, avec une excrétion significative dans la bile. Elles doivent être utilisées avec prudence chez les patients atteints de cirrhose avancée, en raison du risque d’hépatotoxicité. En cas de cirrhose décompensée, il est préférable de choisir des alternatives.
Le métronidazole est métabolisé par le foie via le CYP450, ce qui peut conduire à une accumulation et augmenter le risque de neurotoxicité chez les patients atteints de cirrhose avancée. Une réduction de dose est nécessaire dans ces cas, avec une surveillance attentive pour détecter d’éventuels effets secondaires neurologiques comme la neuropathie ou la confusion.
Les antibiotiques glycopeptidiques, notamment la vancomycine, sont éliminés presque entièrement par voie rénale, avec un métabolisme hépatique négligeable. Leur utilisation nécessite une attention particulière en cas d’insuffisance rénale associée, avec une surveillance des taux sériques pour éviter la néphrotoxicité.
Enfin, la rifampicine est métabolisée par le foie et induit les enzymes hépatiques CYP450, pouvant modifier le métabolisme d’autres médicaments. Son utilisation prolongée doit être évitée chez les patients atteints de cirrhose décompensée, et les interactions médicamenteuses, notamment avec les anticoagulants, doivent être prises en compte.
De plus en plus d’antibiotiques peuvent être adaptés en fonction des dosages sanguin résiduels réalisés. Quand la technique est disponible elle peut être utilisée chez les patients cirrhotiques pour adapter au mieux la posologie.
Connaître les règles de bon usage des psychotropes
Les médicaments agissant sur le système nerveux central (anxiolytiques, sédatifs, antidépresseurs, antipsychotiques, antiépileptiques) sont souvent prescrits aux patients atteints de cirrhose du foie en raison d’une variété de symptômes psychiatriques ou de maladies associées à la cirrhose du foie (28). La plupart des médicaments psychotropes sont lipophiles et sont largement métabolisés par le foie, ce qui implique également une biotransformation par les isoenzymes CYP. Les études cliniques, ainsi que les informations sur les produits, ne fournissent souvent pas de recommandations précises concernant la posologie pour les patients atteints de maladies hépatiques, se limitant à suggérer une réduction de la dose d’un médicament spécifique sans en quantifier la réduction. Dans les études cliniques, les modifications pharmacocinétiques semblent généralement plus importantes chez les patients atteints de cirrhose hépatique que chez ceux souffrant d’hépatite active ou chronique, et elles sont généralement plus marquées à mesure que le score de Child-Pugh augmente.
Tableau 7 : Métabolisme et précaution d’utilisation des antibiotiques (27)
| Médicament | Métabolisme hépatique (Phase I ou II) | Fixation aux protéines (%) | Interaction CYP | Ajustement de dose en cas de cirrhose |
| Azithromycine | Déméthylation (I) | 12-50 | | Aucun |
| Cefotaxime | Désacétylation (II) | 30-50 | | Aucun |
| Ceftriaxone | Non enzymatique, élimination biliaire | 83-96 | | Aucun |
| Ciprofloxacine | Oxydation (I) | 20-40 | Inhibiteur CYP1A2 | Aucun |
| Clarithromycine | Hydroxylation (I), déméthylation (I) | 42-50 | Substrat et inhibiteur CYP3A4 | Aucun |
| Clindamycine | Métabolite sulfoxyde (I), déméthylation (I) | 60-95 | | Child C : 50% réduction |
| Erythromycine | Déméthylation (I) | 73-96 | Substrat et inhibiteur CYP3A4 | Ajustement à prévoir |
| Isoniazide | Acétylation (II) | 4-30 | Inducteur CYP2E1; inhibiteur CYP2C19, CYP1A2, CYP3A4 | Ajustement à prévoir |
| Linzolide | Oxydation (I) | 31 | | Aucun |
| Metronidazole | Hydroxylation (I), oxydation (I), glucuronidation (II) | <20 | Inhibiteur CYP2C9 | Ajustement à prévoir |
| Minocycline | Hydroxylation (I), déméthylation (I) | 76 | | Ajustement à prévoir |
| Moxifloxacine | Glucuronidation (II), sulfatation (II) | 30-50 | | Aucun |
| Nafcilline | Oxydation (I) | 90 | Inducteur CYP1A2 | Ajustement à prévoir |
| Nitrofurantoïne | inconnu | 90 | | Précaution a prendre |
| Norfloxacine | Oxydation (I) | 14 | Inhibiteur CYP1A2 | Aucun |
| Pyrazinamide | Hydrolyse | 5-10 | | Reduire la dose ou éviter l’utilisation |
| Rifampicine | Déacetylation (II), Oxydation (I) | 60-90 | Inducteur CYP3A, CYP 1A2, CYP 2C | Reduire la dose de 50% |
| Sulfamethoxazole- Trimethoprime | Acetylation (II) hydroxylation (I), oxydation (I) | 70, 44 | Substrat et inhibiteur CYP2C9 | Aucun |
| Tigecycline | Glucuronidation (II), N-acétylation (II) | 71-89 | | Aucun |
Tableau 8 : Métabolisme et recommandations d’utilisation des psychotropes
| Médicament | Métabolisme | Effets indésirables hépatiques | Recommandations |
| Bupropion | Hydroxylation (CYP2B6), réduction | Rarement : tests hépatiques anormaux, ictère, hépatite | Commencer avec la dose la plus faible disponible (150 mg). Ajuster selon les effets indésirables dose-dépendants. Éviter chez les patients atteints de cirrhose sévère |
| Sertraline | N-déméthylation (CYP2D6, 2C9, 2B6, 3A4), hydroxylation, glucuronidation | Rarement: hépatite cytolytique, quelques cas d’insuffisance hépatique | Commencer avec 50 % de la dose normale (25 mg). Augmenter seulement après 15 jours. Contre-indiqué chez les patients Child C |
| Chlorpromazine | Hydroxylation (CYP2D6, 1A2), N-déméthylation, sulfoxydation, excrétion biliaire partielle | Risque d’encéphalopathie hépatique, hépatite cholestatique | Éviter chez les patients atteints de cirrhose, si nécéssaire débuter à 25% de la dose. Peut provoquer une encéphalopathie hépatique. |
| Clométhiazole | CYP2A6, 3A4/5, 2B6, glucuronidation | Élévation des transaminases, rarement : jaunisse, hépatite cholestatique | Éviter chez les patients atteints d’insuffisance hépatique sévère. Commencer avec 25 à 50 % de la dose normale. Préférer la voie IV que per os. |
| Hydroxyzine | Métabolisme en cétirizine, 70 % de la dose éliminée par excrétion biliaire | Rarement : hepatite cholestatique, ictère | Commencer avec 50 % de la dose normale. Ajuster en fonction des effets indésirables dose-dépendants anticholinergique, somnolence). |
| Quétiapine | Désalkylation, hydroxylation (CYP3A4, 2D6), sulfoxydation, glucuronidation | Rarement : ictère, élévations transitoires des transaminases | Commencer avec 25 mg par jour, augmenter progressivement en fonction des réactions cliniques. Éviter chez les patients atteints de cirrhose décompensée |
| Buspirone | Désalkylation, hydroxylation (CYP3A4), glucuronidation | Rarement : somnolence, asthénie | Commencer avec une dose réduite de 5 mg par jour chez les patients atteints d’insuffisance hépatique. Éviter chez les patients atteints d’insuffisance hépatique sévère. |
| Zaleplon | Oxydase aldéhydique, désalkylation (CYP3A4), glucuronidation | | Commencer avec la dose la plus faible (5 mg) chez les patients atteints d’insuffisance hépatique légère à modérée. Éviter chez les patients atteints d’insuffisance hépatique sévère. |
L’ajustement des doses est donc particulièrement important chez les patients atteints de cirrhose. La combinaison des résultats des études pharmacocinétiques et des estimations basées sur l’extraction hépatique peut être utile pour aider les cliniciens à choisir une posologie adaptée pour ces patients. Cependant, les essais cliniques prospectifs testant la pertinence de telles recommandations posologiques font largement défaut, et ces propositions ne doivent donc pas être interprétées comme des lignes directrices définitives en matière de posologie. Les recommandations posologiques basées sur l’extraction hépatique et/ou la biodisponibilité d’un médicament sont généralement en bon accord avec les données issues des études pharmacocinétiques chez les patients atteints de cirrhose hépatique. La classification des médicaments en fonction de leur extraction hépatique constitue donc une approche utile pour l’ajustement des doses chez ces patients, en l’absence d’études cliniques appropriées. Ce ne sont pas seulement les modifications de la pharmacocinétique du médicament principal et/ou de ses métabolites actifs qui doivent être prises en compte, mais également la situation clinique du patient et l’indice thérapeutique du médicament, qui peuvent influencer le choix de la posologie pour certains traitements.
En conclusion, la prescription de traitements chez les patients cirrhotiques, et particulièrement ceux classés Child-Pugh C, requiert une vigilance accrue en raison des altérations du métabolisme hépatique et de l’élimination des médicaments. Les ajustements posologiques doivent être basés sur une évaluation précise de la sévérité de la cirrhose et des caractéristiques pharmacocinétiques des médicaments. Les patients Child-Pugh C présentent un risque particulièrement élevé de toxicité, nécessitant une approche individualisée et une surveillance étroite. Cependant, les données pharmacologiques restent limitées chez cette population, du fait de leur exclusion fréquente des essais cliniques.
Références
- Almazroo OA, Miah MK, Venkataramanan Drug Metabolism in the Liver. Clin Liver Dis. 2017 Feb;21(1):1–20.
- Dietrich CG, Götze O, Geier Molecular changes in hepatic metabolism and transport in cirrhosis and their functional importance. World J Gastroenterol. 2016 Jan 7;22(1):72–88.
- Thakkar N, Slizgi JR, Brouwer Effect of Liver Disease on Hepatic Transporter Expression and Function. J Pharm Sci. 2017 Sep;106(9):2282–94.
- El-Khateeb E, Darwich AS, Achour B, Athwal V, Rostami-Hodjegan Review article: time to revisit Child-Pugh score as the basis for predicting drug clearance in hepatic impairment. Aliment Pharmacol Ther. 2021 Aug;54(4):388–401.
- Duthaler U, Bachmann F, Suenderhauf C, Grandinetti T, Pfefferkorn F, Haschke M, et Liver Cirrhosis Affects the Pharmacokinetics of the Six Substrates of the Basel Phenotyping Cocktail Differently. Clin Pharmacokinet. 2022 Jul;61(7):1039–55.
- El-Khateeb E, Achour B, Al-Majdoub ZM, Barber J, Rostami-Hodjegan Non-uniformity of Changes in Drug-Metabolizing Enzymes and Transporters in Liver Cirrhosis: Implications for Drug Dosage Adjustment. Mol Pharm. 2021 Sep 6;18(9):3563–77.
- Delcò F, Tchambaz L, Schlienger R, Drewe J, Krähenbühl Dose adjustment in patients with liver disease. Drug Saf. 2005;28(6):529–45.
- García-Cortés M, García-García Management of Pharmacologic Adverse Effects in Advanced Liver Disease. Clin Drug Investig. 2022 Jun;42(Suppl 1):33–8.
- Frye RF, Zgheib NK, Matzke GR, Chaves-Gnecco D, Rabinovitz M, Shaikh OS, et Liver disease selectively modulates cytochrome P450– mediated metabolism. Clin Pharmacol Ther. 2006 Sep;80(3):235–45.
- Andrade RJ, Aithal GP, Björnsson ES, Kaplowitz N, Kullak-Ublick GA, Larrey D, et al. EASL Clinical Practice Guidelines: Drug-induced liver J Hepatol [Internet]. 2019 Mar 26 [cited 2019 Apr 14];0(0). Available from: https://www.journal-of-hepatology.eu/article/S0168- 8278(19)30129-1/abstract
- Chandok N, Watt Pain management in the cirrhotic patient: the clinical challenge. Mayo Clin Proc. 2010 May;85(5):451–8.
- Khalid SK, Lane J, Navarro V, Garcia-Tsao Use of over-the-counter analgesics is not associated with acute decompensation in patients with cirrhosis. Clin Gastroenterol Hepatol Off Clin Pract J Am Gastroenterol Assoc. 2009 Sep;7(9):994–9; quiz 913–4.
- Schweighardt AE, Juba A Systematic Review of the Evidence Behind Use of Reduced Doses of Acetaminophen in Chronic Liver Disease. J Pain Palliat Care Pharmacother. 2018 Dec;32(4):226–39.
- De Lédinghen V, Heresbach D, Fourdan O, Bernard P, Liebaert-Bories MP, Nousbaum JB, et al. Anti-inflammatory drugs and variceal bleeding: a case-control study. Gut. 1999 Feb;44(2):270–3.
- Rakoski M, Goyal P, Spencer-Safier M, Weissman J, Mohr G, Volk Pain management in patients with cirrhosis. Clin Liver Dis. 2018 Jun;11(6):135–40.
- Dwyer JP, Jayasekera C, Nicoll Analgesia for the cirrhotic patient: a literature review and recommendations. J Gastroenterol Hepatol. 2014;29(7):1356–60.
- Pereira Portela C, Gautier LA, Zermatten MG, Fraga M, Moradpour D, Bertaggia Calderara D, et Direct oral anticoagulants in cirrhosis: Rationale and current evidence. JHEP Rep Innov Hepatol. 2024 Aug;6(8):101116.
- Ma J, Chalasani NP, Schwantes-An L, Björnsson Review article: the safety of anticoagulants and antiplatelet agents in patients with cirrhosis. Aliment Pharmacol Ther. 2023 Jan;57(1):52–71.
- Smith FC, Stocker SL, Danta M, Carland JE, Kumar SS, Liu Z, et The safety and pharmacokinetics of metformin in patients with chronic liver disease. Aliment Pharmacol Ther. 2020 Mar;51(5):565–75.
- Shaffer LR, Mahmud Statins in Cirrhosis: Hope or Hype? J Clin Exp Hepatol. 2023;13(6):1032–46.
- Pose E, Trebicka J, Mookerjee RP, Angeli P, Ginès Statins: Old drugs as new therapy for liver diseases? J Hepatol. 2019 Jan;70(1):194–202.
- Law M, Rudnicka Statin safety: a systematic review. Am J Cardiol. 2006 Apr 17;97(8A):52C-60C.
- Abraldes JG, Villanueva C, Aracil C, Turnes J, Hernandez-Guerra M, Genesca J, et Addition of Simvastatin to Standard Therapy for the Prevention of Variceal Rebleeding Does Not Reduce Rebleeding but Increases Survival in Patients With Cirrhosis. Gastroenterology. 2016 May;150(5):1160-1170.e3.
- Pose E, Napoleone L, Amin A, Campion D, Jimenez C, Piano S, et al. Safety of two different doses of simvastatin plus rifaximin in decompensated cirrhosis (LIVERHOPE-SAFETY): a randomised, double-blind, placebo-controlled, phase 2 Lancet Gastroenterol Hepatol. 2020 Jan;5(1):31–41.
- Ao X, Zeng Y, Wang X, Fan Letter: Fibrates may be safe and effective in patients with primary biliary cholangitis and decompensated cirrhosis. Aliment Pharmacol Ther. 2024;60(1):105–6.
- Crocombe D, O’Brien Antimicrobial prophylaxis in decompensated cirrhosis: friend or foe? Hepatol Commun. 2023 Sep 1;7(9):e0228.
- Halilovic J, Heintz Antibiotic dosing in cirrhosis. Am J Health-Syst Pharm AJHP Off J Am Soc Health-Syst Pharm. 2014 Oct 1;71(19):1621–34.
- Lucena MI, Andrade RJ, Tognoni G, Hidalgo R, Sanchez de la Cuesta F, the Spanish Collaborative Study Group on Therapeutic Management in Liver Diseases. Drug use for non-hepatic associated conditions in patients with liver cirrhosis. Eur J Clin Pharmacol. 2003 May 1;59(1):71–6.