Toute reproduction ou réécriture, totale ou partielle, sans l’accord préalable écrit de la FMC HGE est interdite.
Objectifs pédagogiques
Savoir évoquer devant une stéatose une autre cause que l’alcool et le dysmétabolisme (MetAld)
Connaître les causes des stéatoses non MetAld
Savoir confirmer le diagnostic d’une stéatose non MetAld
Connaître les modalités thérapeutiques
Les points forts
Face à une stéatose « inexpliquée », il convient en premier lieu d’éliminer une consommation d’alcool mal évaluée et une présentation atypique d’une stéatose liée au dysmétabolisme (MASLD du sujet mince).
La démarche est ensuite guidée par un interrogatoire détaillé et l’analyse du contexte global (dénutrition, perte de poids très rapide post chirurgie bariatrique, nutrition parentérale, endocrinopathie…).
L’analyse du bilan lipidique sanguin peut orienter vers des causes métaboliques génétiques rares (hypobétalipoprotéinémie, déficit en lipase acide lysosomale…).
La prise de certains médicaments (tamoxifène, corticothérapie chronique, certaines chimiothérapies, antirétroviraux de première génération), peut favoriser une stéatose hépatique, macro et/ou microvésiculaire.
Le diagnostic précis de ces causes secondaires de stéatose hépatique doit être fait pour permettre d’initier un traitement spécifique. En l’absence de diagnostic étiologique, le suivi par des tests non invasifs de la fibrose hépatique est utile.
ABHD5 : Alpha/Beta Hydrolase Domain-Containing 5 (gène, CGI-58) ; ADRA2A : Adrenergic receptor, Alpha-2A subtype (gène) ; AGPAT2 : 1-acylglycerol-3-phosphate O-acyltransferase 2 (gène) ; AINS : Anti-inflammatoires non stéroïdiens ; AKT2 : v-akt murine thymoma viral oncogene homolog 2 (gène) ; ALD : Alcohol-related Liver Disease (maladie du foie liée à l’alcool) ; ALDOB : Aldolase B (gène) ; ANGPTL3 : Angiopoietin-like 3 (gène) ; ANSES : Agence nationale de sécurité sanitaire de l’alimentation, de l’environnement et du travail ; ApoA : Apolipoprotéine A ; ApoB / APOB : Apolipoprotéine B (APOB = gène) ; ATP7B : ATPase transportant le cuivre (gène de la maladie de Wilson), BPA : Bisphénol A ; BSCL2 : Berardinelli-Seip congenital lipodystrophy 2 (gène) ; CAP : (Continuous) Controlled Attenuation Parameter (paramètre d’atténuation contrôlée du FibroScan) ; CASH : Chemotherapy-Associated Steatohepatitis ; CAV1 : Caveolin-1 (gène) ; CAVIN1 : Caveolae-Associated Protein 1 (gène) ; CGI-58 : Comparative Gene Identification-58 (cofacteur, associé à ABHD5) ; CHC : Carcinome hépatocellulaire ; CIDEC : Cell Death-Inducing DFFA-Like Effector C (gène) ; CPK : Créatine phosphokinase (créatine kinase) ; CSAPA : Centre de soins, d’accompagnement et de prévention en addictologie ; CTLN2 : Citrullinémie de type 2 ; dB/m : Décibel par mètre (unité utilisée pour le CAP) ; DIS : Drug-Induced Steatosis ; DISH : Drug-Induced Steatohepatitis ; ELF : Enhanced Liver Fibrosis (test sanguin ELF®) ; F : Femme ; FIB-4 : Fibrosis-4 index ; H : Homme ; HAART : Highly Active Antiretroviral Therapy (multithérapie antirétrovirale) ; HDL : High-Density Lipoprotein (cholestérol HDL) ; HELLP : Hemolysis, Elevated Liver enzymes, Low Platelet count (syndrome HELLP) ; HFE : Gène HFE (hémochromatose) ; HSD17B13 : Hydroxysteroid 17-beta dehydrogenase 13 (gène) ; IFALD : Intestinal Failure-Associated Liver Disease ; IMC : Indice de masse corporelle ; LAL / LAL-D : Lysosomal Acid Lipase / déficit en lipase acide lysosomale ; LDL : Low-Density Lipoprotein (cholestérol LDL) ; LIPE : Hormone-Sensitive Lipase (gène) ; LIPA : Lipase A (gène ; déficit en LAL) ; LMNA : Lamin A/C (gène) ; MAF : Minor Allele Frequency (fréquence allélique mineure) ; MASLD : Metabolic dysfunction-Associated Steatotic Liver Disease ; MASH : Metabolic dysfunction-Associated Steatohepatitis ; MBOAT7 : Membrane Bound O-Acyltransferase Domain-Containing 7 (gène) ; MetALD : Entité “mixte” : MASLD + consommation d’alcool ; MTTP : Microsomal Triglyceride Transfer Protein (gène) ; NHANES : National Health and Nutrition Examination Survey (cohorte américaine) ; NICCD : Neonatal Intrahepatic Cholestasis caused by Citrin Deficiency ; NNRTI : Non-Nucleoside Reverse Transcriptase Inhibitor (inhibiteur non nucléosidique) ; NRTI : Nucleoside Reverse Transcriptase Inhibitor (inhibiteur nucléosidique) ; OR : Odds Ratio ; PCSK9 : Proprotein Convertase Subtilisin/ Kexin type 9 (gène) ; PCB : Polychlorobiphényles ; PDFF : Proton Density Fat Fraction (séquence IRM de quantification de la graisse) ; PFAS : Substances per- et polyfluoroalkylées ; Pi*Z : Variant “Z” de l’alpha-1 antitrypsine ; PLIN1 : Perilipin-1 (gène) ; PPARG : Peroxisome Proliferator-Activated Receptor Gamma (gène) ; RUCAM : Roussel-Uclaf Causality Assessment Method (imputabilité médicamenteuse) ; SERPINA1 : gène de l’alpha-1 antitrypsine ; SHAG : Stéatose hépatique aiguë gravidique ; SLC25A13 : Solute Carrier Family 25 Member 13 (gène, citrine) ; SOPK : Syndrome des ovaires polykystiques ; TASH : Toxicant-Associated Steatosis or Steatohepatitis ; TM6SF2 : Transmembrane 6 Superfamily Member 2 (gène) ; TSH : Thyroid-Stimulating Hormone (thyréostimuline) ; VHB : Virus de l’hépatite B ; VHC : Virus de l’hépatite C ; VIH : Virus de l’immunodéficience humaine.
Résumé
Des stéatoses hépatiques non liées à l’alcool et non liées au dysmétabolisme se verraient dans 0,03 à 0,6 % de la population générale. Les données de l’interrogatoire et le contexte clinique global peuvent orienter vers les causes les plus fréquentes de ces stéatoses hépatiques secondaires, notamment médicamenteuses. Certaines anomalies du bilan lipidique, présentes en dehors d’un contexte dysmétabolique ou lié à l’alcool, doivent faire rechercher des causes génétiques perturbant le métabolisme lipidique. Lorsqu’une cause est trouvée, un traitement spécifique, s’il existe, doit être mis en place. En cas de stéatose de cause inconnue, l’évaluation initiale de la fibrose hépatique et un suivi régulier sont utiles.
Introduction
Les stéatoses hépatiques correspondent à une surcharge lipidique dans les hépatocytes. Classiquement, l’histologie hépatique permet de distinguer les stéatoses macrovacuolaires (grosse gouttelette de triglycérides refoulant le noyau de l’hépatocyte), des stéatoses micro vésiculaires (petites gouttelettes de triglycérides ne refoulant pas le noyau de l’hépatocyte). Les mécanismes physiopathologiques altérant le métabolisme lipidique hépatocytaire responsables de ces 2 types de stéatose peuvent être soit communs, soit spécifiques (dysfonctions mitochondriales rencontrées dans certaines stéatoses micro vésiculaires).
Lorsque la stéatose macrovacuolaire touche plus de 20 à 30 % des hépatocytes sur une biopsie hépatique, elle est généralement visible à l’analyse du parenchyme hépatique lors d’une échographie abdominale ou d’une imagerie en coupes scanographique ou par résonance magnétique nucléaire. À l’inverse, les stéatoses hépatiques débutantes et souvent les stéatoses purement microvésiculaires sont non visibles. De nouveaux examens permettent désormais une détection et une quantification beaucoup plus précises. L’examen de référence est l’imagerie par résonance magnétique nucléaire en séquence PDFF (au seuil de 5 à 10 % pour diagnostiquer la stéatose histologique S1 ; 11 à 18 % pour la stéatose S2 ; 16 à 23 % pour la stéatose S3), et dans une moindre mesure, mais beaucoup plus facilement accessible et répétable, le (continuous) Controlled Attenuation Parameter (CAP) donné par le FibroScan (Echosens), avec des seuils d’environ 250 à 268 dB/m pour la stéatose S1, 268 à 280 dB/m pour la stéatose S2, et supérieur à 280 dB/m pour la stéatose S3 (1-3).
Chez l’adulte, la démarche diagnostique devant une stéatose hépatique, découverte lors d’un examen d’imagerie abdominale, est fréquente et souvent rapide, compte tenu de la relative facilité de diagnostiquer les trois causes largement majoritaires : la maladie du foie liée à l’alcool [Alcohol-related Liver Disease (ALD)], la stéatose hépatique dysmétabolique [Metabolic dysfunction-Associated Steatotic Liver Disease (MASLD)], et l’entité MetALD définie depuis 2023 (MASLD et consommation d’alcool allant de 4 à 6 verres/j chez l’homme et de 3 à 5 verres/j chez la femme) (4). Ces
3 entités correspondent chacune à un continuum de lésions histologiques allant de la stéatose hépatique simple, à la stéatohépatite, la stéatohépatite avec fibrose, la cirrhose et les complications de la cirrhose. Si ces causes principales sont absentes, commence alors une démarche diagnostique, parfois difficile, à la recherche d’une cause de stéatose hépatique secondaire (5). En population générale américaine (cohorte NHANES étudiée de 2017 à 2020), ces stéatoses étaient rares (0,03 à 0,6 % de la population générale selon le seuil de CAP utilisé pour la détection de la stéatose) (6). La fréquence de ces atteintes peut probablement être plus élevée lors d’une consultation d’hépatologie en centre de recours.
Une enquête clinique minutieuse basée sur un interrogatoire détaillé et un examen physique général, ainsi que l’analyse du bilan lipidique donné par une « exploration d’une anomalie lipidique » peuvent orienter vers une cause. En cas de négativité, des examens de 2e ligne peuvent être proposés. La prise en charge optimale de certaines maladies ainsi diagnostiquées peut nécessiter l’aide d’autres spécialistes.
Quelques situations très particulières sont associées à un cortège d’anomalies cliniques et paracliniques touchant le foie et parfois d’autres organes. Les anomalies hépatiques peuvent être associées à une stéatose hépatique aiguë micro vésiculaire, généralement uniquement diagnostiquée lors d’une éventuelle biopsie hépatique. Certaines de ces situations sont des urgences thérapeutiques (5).
Éliminer une présentation atypique d’une MASLD, d’une MetALD ou d’une maladie du foie liée à l’alcool
Chez un adulte, avant d’entamer une démarche à la recherche d’une cause rare d’une stéatose hépatique secondaire, il est important d’éliminer une présentation atypique d’une MASLD, d’une MetALD ou d’une maladie du foie liée à l’alcool.
La plupart des personnes ayant une MASLD sont faciles à reconnaître parfois à la simple inspection, du fait d’un fort surpoids ou d’une obésité androïde marquée. Cependant, 10 à 15 % des personnes avec une MASLD ont un phénotype atypique marqué par un discret surpoids, voire un tour de taille normal. Cette entité, bien décrite depuis 15 ans, peut être difficile à diagnostiquer (7). Elle présente autant, voire plus de risque évolutif vers une fibrose hépatique avancée et des complications hépatiques, ainsi qu’un risque accru d’évènements cardio-vasculaires et de cancers extra-hépatiques (8). La définition actuelle de la MASLD (présence d’une stéatose hépatique et au moins un élément de risque cardio-métabolique parmi les éléments du syndrome métabolique : augmentation du tour de taille, élévation de la pression artérielle ou la prise d’un traitement anti-hypertenseur, élévation de la glycémie à jeun ou la prise d’un médicament anti-diabétique, élévation de la triglycéridémie ou la prise d’un médicament hypolipémiant spécifique, ou la baisse du HDL-cholestérol ou la prise d’un médicament hypolipémiant spécifique) permet de diagnostiquer ces patients (4). L’interrogatoire sur l’histoire pondérale retrouve souvent une prise de poids d’au moins une dizaine de kilos entre l’âge de 20 ans et le moment de la consultation pour la stéatose hépatique. La prise de poids s’est alors généralement faite au niveau abdominal, même si le tour de taille reste inférieur ou proche des seuils définis selon les origines ethniques [Européens : ≥ 94 cm chez l’homme (H), 80 cm chez la femme (F) ; Américains : ≥ 102 cm (H) ≥ 88 cm (F) ; Asie du Sud et Chine ≥ 90 cm (H), 80 cm (F), Japon ≥ 85 cm (H), 90 cm (F)]. La prise en charge de ces patients est globalement identique à celle proposée pour les patients ayant une MASLD classique. La perte de poids peut être bénéfique chez eux et est recommandée, même si les données de la littérature sont encore limitées (3,9).
Plus rarement, une maladie du foie liée à l’alcool peut avoir une présentation trompeuse. Le piège le plus fréquent est le fait d’un interrogatoire trop succinct ou basé sur des questions imprécises ou trop fermées, auxquelles, de bonne foi le patient répond en induisant en erreur le clinicien. Ainsi, la question « Buvez-vous de l’alcool ? » est souvent comprise comme une interrogation sur la prise d’alcool fort (spiritueux). Ainsi, l’interrogatoire doit être méthodique et notamment explorer les différents types de boissons alcoolisées (vin, bière, spiritueux) ainsi que les quantités et les rythmes d’alcoolisation. Il s’agit par exemple de reconnaître les alcoolisations intermittentes mais massives [alcoolisation ponctuelle importante (≥ 6 verres en une occasion chez l’adulte et ≥ 5 verres en une occasion chez l’adolescent)] des soirées de week-end qui peuvent être « oubliées » ou minimisées par certains patients par ailleurs totalement sobres en semaine. Les américains distinguent le « binge drinking » (≥ 4 verres chez la femme et ≥ 5 verres chez l’homme en une occasion, au moins une fois par mois), le « heavy use » (binge drinking au moins 5 fois dans le mois) et le « high intensity drinking » (≥ 10 verres en une occasion dans les 15 derniers jours).
Un autre piège est la situation, rare, d’un patient avec une maladie du foie liée à l’alcool qui présente un déni farouche de toute consommation d’alcool. Ce déni du patient est parfois confirmé par une personne accompagnante (dite « de confiance »). Celle-ci peut, de bonne foi, ignorer une consommation cachée par l’accompagné(e) ou plus rarement être complice du déni. La présence d’anomalies biologiques (hépatiques ou à l’hémogramme) notables est classique au cours de l’intoxication chronique. La sévérité de l’évolution hépatique lorsque le patient est suivi, ou une histologie hépatique compatible lorsqu’une biopsie est faite, et surtout les nouveaux marqueurs de la consommation d’alcool (phosphatidyléthanol sérique, éthylglucuronide capillaire) peuvent redresser le diagnostic. Ces tests ont globalement une bonne sensibilité et une bonne spécificité pour diagnostiquer une consommation en alcool, notamment « à risque » [> 3 verres/j (H), > 2 verres/j (F)] et d’excellentes valeurs prédictives négatives. Si ces outils peuvent offrir une opportunité d’ouvrir un dialogue sur la consommation d’alcool en population de soins primaires, la confrontation d’un patient cachant une consommation d’alcool avec le résultat de l’un de ses tests positifs ou une alcoolémie « surprise » positive peut déstabiliser le patient alors « démasqué » (10). La relation médecin/patient peut alors être délicate, avec des réactions de colère ou de déni persistante et un risque de perte de vue. Une approche multidisciplinaire avec une équipe d’un centre de soin, d’accompagnement et de prévention en addictologie (CSAPA) est adéquate si l’adhésion du patient est finalement obtenue.
Faire une enquête médicamenteuse exhaustive
De nombreux médicaments prescrits par un médecin ou parfois achetés sans ordonnance sont classiquement associés à la présence d’une stéatose hépatique (tableau 1) (11,12). Les maladies hépatiques stéatosiques liées aux médicaments sont actuellement nommées en anglais drug-induced steatosis (DIS) ou drug-induced steatohepatitis (DISH). Les maladies hépatiques stéatosiques liées à des agents de chimiothérapies anti-cancéreuses sont nommées chemotherapy-associated steatohepatitis (CASH) (13). L’enquête médicamenteuse fait partie de l’examen clinique de base de tout médecin prenant en charge une personne avec une stéatose hépatique accompagnée ou non d’une anomalie du bilan hépatique. La prise chronique de corticoïdes, médicaments très classiquement associés à une stéatose hépatique, est souvent retrouvée chez des personnes ayant aussi eu une histoire de prise pondérale parfois associée à une MASLD (11,12). L’archétype sont les personnes avec des maladies rhumatologiques articulaires inflammatoires (maladie rhumatoïde et spondylarthropathie ankylosante) qui ont des douleurs polyarticulaires limitant durablement l’activité physique et nécessitant parfois des prises régulières de corticoïdes. Certaines personnes avec des douleurs chroniques peuvent également présenter un syndrome dépressif qui participe à limiter durablement le niveau d’activité physique régulière. À côté des médicaments classiquement associés à une stéatose hépatique (tableau 1), certains médicaments neuroleptiques ont des effets orexigènes et sont parfois associés à un état d’apathie limitant l’activité physique. L’effet conjugué d’une augmentation de la prise alimentaire et d’une faible dépense énergétique participe à une prise de poids et à l’apparition d’une stéatose hépatique.
Mécanismes
Type de stéatose à la biopsie hépatique
Possibilité d’une aggravation d’une MASLD coexistence
Traitements du VIH : NRTIs (didanosine, stavudine, zidovudine)NNRTI (efavirenz)
1, 2, 3
MicroS, macroS
Oui
Médicaments du VIH de première génération, actuellement inusités.
Traitements du VIH : anti-protéase boostée au ritonavir
Dans le cadre d’un syndrome lipodystrophique
Méthotrexate
2
MacroS, stéatohépatite
Oui
Imputabilité finalement faible. Terrain de MASLD très souvent présent
Tétracycline
2, 3
MicroS
Méfloquine
MacroS
Acide valproïque
2
MicroS
Cocaïne
2
MicroS
VitamineA
MicroS
Tableau 1 : Principaux médicaments associés à la présence d’une stéatose hépatique [adapté d’après (11,12,16,24)] 1 : augmentation de la synthèse des acides gras 2 : diminution de la béta-oxydation des acides gras 3 : Diminution de l’export des lipoprotéines 4 : augmentation de la mobilisation et de la capture des acides gras AINS : Anti-inflammatoire non stéroïdiens, HAART : Highly Active Antiretroviral Therapy ; MicroS : Stéatose microvésiculaire, macroS : stéatose macrovacuolaire, NRTI : inhibiteur nucléosidique de la transcriptase inverse, NNRTI : inhibiteur non nucléosidique de la transcriptase inverse.
Le méthotrexate était classiquement associé à l’apparition d’une stéatose, d’une stéatohépatite, d’une fibrose et d’une cirrhose. Cependant l’analyse actualisée et attentive de la littérature suggère une fréquente coexistence d’une MASLD parfois au stade de MASH fibrosante ou de cirrhose MASH avec une toxicité propre du méthotrexate nulle ou modérée (14).
La démarche diagnostique peut se baser sur le RUCAM (Roussel-Uclaf Causality Assessment Method) avec une étude de l’imputabilité médicamenteuse intrinsèque et extrinsèque. Cela implique d’éliminer les autres causes classiques de stéatose hépatique (MASLD, MetALD, maladie du foie liée à l’alcool, et les autres causes de stéatose hépatique secondaire), d’évaluer la chronologie d’usage du médicament et la date supposée d’apparition de la stéatose hépatique (généralement impossible à connaître avec précision), et d’effectuer une recherche bibliographique. L’aide d’un service de pharmacovigilance peut être utile (15).
Dans la situation de stéatose hépatique liée ou favorisée par un médicament, la question de l’arrêt de celui-ci se pose. Cependant, certains médicaments seront indispensables au patient et ne pourront pas être arrêtés. L’avis du médecin spécialiste prescripteur du médicament en question sera utile pour évaluer les possibilités de réduction de dose, d’arrêt ou de substitution.
En l’absence de possibilité d’arrêt du médicament en question, l’évaluation initiale puis une surveillance, tous les 1 à 3 ans, avec des tests de fibrose hépatique non-invasifs (FibroScan), sont pertinentes.
Une enquête doit être faite également à la recherche de la prise d’éventuels compléments alimentaires, ou d’herbes médicinales ou de produits de naturopathie. L’aide de la nutrivigilance organisée par l’ANSES peut être utile ([email protected]). Une déclaration en ligne est facilement réalisée (https://www.nutrivigilance-anses.fr/nutri/teledeclarer#!).
La recherche d’une infection par le VIH et des traitements pris actuellement et dans le passé est importante. En effet, l’infection par le VIH elle-même, mais surtout les premiers inhibiteurs nucléosidiques de la transcriptase inverse (zidovudine, stavudine, didanoside), et, de façon moins documentée, l’efavirenz, un inhibiteur non nucléosidique de la transcriptase inverse, ont été associés à la présence d’une stéatose hépatique (16). Ces effets toxiques seraient en partie liés à une toxicité mitochondriale et en partie à une altération hépatocytaire du métabolisme des lipides. De plus, certaines anti-protéases ont été associées à la présence d’une lipodystrophie du VIH. Cette lipodystrophie est très fréquemment associée à des dépôts graisseux viscéraux ectopiques dont une stéatose hépatique. Le syndrome de lipodystrophie du VIH peut comporter une lipoatrophie (du visage et des membres), une lipohypertrophie abdominale (augmentation de la graisse viscérale) et une lipomatose de la région cervico-dorsale (bosse de bison). Vingt-trois à 70 % des personnes vivant avec le VIH présenteraient une lipodystrophie (17). Si les molécules les plus inductrices de stéatose hépatique et/ou de lipodystrophies ne sont plus utilisées, beaucoup des patients traités depuis 30 ans les ont reçues (12). Les traitements plus récents du VIH (ténofovir, inhibiteurs d’entrée (maraviroc) ou les inhibiteurs d’intégrase (raltegravir, dolutegravir)) induisent moins d’effets secondaires (16).
Des arguments sont présents dans la littérature pour souligner que la présence concomitante d’une MASLD peut être observée et favoriserait la toxicité de certains de ces médicaments (18). Une surveillance de la fibrose hépatique à l’aide des tests non invasifs (FibroScan) est utile.
À l’interrogatoire, évaluer le contexte global et évoquer certaines causes classiques de stéatose hépatique
Comme toujours, un interrogatoire détaillé et la prise en compte du contexte global du patient peut permettre d’évoquer des causes rares mais classiques de stéatose hépatique secondaire.
Stéatose hépatique et infection par le VHC de génotype 3
La coexistence d’une MASLD et d’une infection par le virus de l’Hépatite C (VHC) est possible. Cependant le VHC de génotype 3 a été rapporté comme spécifiquement associé à la présence d’une stéatose hépatique virale qui disparaît lors de la guérison après un traitement anti viral spécifique (5). Cela participe à justifier l’intérêt de toujours effectuer une recherche sérologique des virus ayant des modes de contamination communs (VHC, VHB et VIH) chez une personne consultant pour une stéatose hépatique et/ou une perturbation du bilan hépatique.
Stéatose hépatique et malnutritions
Une situation vue en service de nutrition est la stéatose hépatique liée à une nutrition parentérale composée d’apports lipidiques excessifs ou mal équilibrés chez des patients ayant une insuffisance intestinale. La stéatose hépatique (micro ou micro et macrovacuolaire) se voit alors dans 65 % des présentations de cet effet secondaire appelé « intestinal failure-associated liver disease (IFALD) ». L’autre forme d’IFALD, est une maladie cholestatique hépatique. En cas d’IFALD, une fibrose puis une cirrhose et une décompensation peuvent survenir (19). Une transplantation hépatique peut alors se discuter. Les médecins nutritionnistes sont bien formés à ces complications et essayent toujours d’adapter de façon curative ou mieux préventive, la composition et la quantité de la nutrition parentérale aux besoins du patient. Dans le cadre de la stéatose hépatique post-nutrition parentérale, le changement de la composition du mélange nutritif avec un apport diminué et qualitativement modifié en lipides, voire un sevrage de la nutrition parentérale, quand cela est possible, permet de diminuer, voire corriger, les anomalies hépatiques (5).
Dans certaines situations de grande dénutrition – notamment l’anorexie mentale sévère, la pauvreté extrême avec marasme ou kwashiorkor – une stéatose hépatique peut survenir (5). Une prise en charge spécifique est alors nécessaire (réalimentation progressive, prévention du syndrome de renutrition inappropriée, et surveillance hépatique et nutritionnelle).
Une autre situation archétypale est la stéatose apparaissant ou s’aggravant dans les suites d’une perte de poids majeure et brutale liée à la réalisation récente d’une chirurgie bariatrique/métabolique (5). Plusieurs séries anciennes de cas rapportent des stéatoses et des stéatohépatites, voire des insuffisances hépatocellulaires, ayant parfois mené à d’exceptionnelles transplantations hépatiques, dans les suites de chirurgie bariatrique. Cela concernait principalement des techniques historiques de dérivation extrême du flux intestinal (bypass jéjuno-iléal). Cette complication est devenue très rare à une époque où les chirurgiens digestifs ont développé depuis 30 ans des techniques fiables (gastric-by-pass et sleeve gastrectomy) (20). Dans l’éventualité de cette situation, une nutrition entérale doit être débutée avec une prévention soigneuse d’un syndrome de renutrition inappropriée (supplémentation en polyvitamines, phosphore, magnésium et oligo-éléments).
La pancréatectomie totale peut également se compliquer d’une stéatose hépatique chez 37 à 50 % des patients. Celle-ci est favorisée par une dénutrition et une supplémentation en enzymes pancréatiques insuffisante. Un support nutritionnel et une supplémentation en enzymes pancréatiques adéquats constituent les traitements de cette situation (5).
Stéatose hépatique et endocrinopathies
De nombreuses endocrinopathies peuvent aggraver une MASLD ou participer à une stéatose hépatique secondaire (5).
Dans une série de 879 patients avec un panhypopituitarisme, une obésité hypothalamique ou un craniopharyngiome, 2,3 % des patients avaient une stéatose hépatique secondaire. Certains patients avaient une progression en environ 6 ans vers une cirrhose. Une majorité de patients avec un hypopituitarisme développait une obésité, une intolérance au glucose ou un diabète, une hypertrigycéridémie, voire une hypercholestérolémie. Ces anomalies seraient la conséquence d’une carence en hormone de croissance (21).
Une hypothyroïdie franche ou minime a été liée à une MASLD et est souvent associée à des éléments du syndrome métabolique. Le développement de la stéatose hépatique serait lié à un effet direct de la TSH sur le métabolisme des triglycérides, ainsi qu’à une diminution de la production d’insuline par les cellules béta pancréatiques et à une dérépression de la lipolyse du tissu adipeux qui augmente le flux d’acides gras libres vers le foie (5). L’efficacité du resmetirom, qui est un agoniste des récepteurs béta des hormones thyroïdiennes, présents surtout au niveau hépatique, testé chez des patients avec une MASH fibrosante sans cirrhose, pour la résolution de la MASH sans aggravation de la fibrose (groupe 100 mg : 30 % versus 10 % pour le placebo, p< 0,001) et l’amélioration de la fibrose sans aggravation de la MASH (groupe 100 mg : 26 % versus 14 % pour le placebo, p< 0,001) illustre l’importance de cette voie, au moins chez une partie des patients, au niveau du métabolisme lipidique et énergétique des hépatocytes (22).
Le syndrome des ovaires polykystiques (SOPK) est fortement associé à l’insulinorésistance et au syndrome métabolique. Il n’est donc pas étonnant que la MASLD soit fréquemment retrouvée chez des patientes atteintes d’un SOPK. La prévalence augmentée de la stéatose hépatique chez des jeunes patientes atteintes, même en l’absence d’obésité ou de syndrome métabolique, illustre l’impact d’autres facteurs dans cette maladie, comme l’hyperandrogénie. En effet, l’hyperandrogénie et l’élévation de la testostéronémie étaient significativement associées à la stéatose hépatique indépendamment de la présence d’une insulino-résistance. Le SOPK serait donc associé à la fois à une MASLD dans un contexte d’insulinorésistance et à une stéatose hépatique secondaire en cas d’hyperandrogénie (5).
Chez l’homme, de faibles concentrations de testostérone sont associées à une prévalence accrue de stéatose hépatique, une relation qui persiste après ajustement sur l’IMC et certains facteurs métaboliques, bien que l’indépendance vis-à-vis de l’insulinorésistance reste débattue (5).
Ainsi, l’excès d’hormones androgènes chez la femme au cours d’un SOPK ou la carence d’hormones androgènes chez l’homme seraient associés à la fois à des situations de MASLD et à des stéatoses hépatiques secondaires.
La suspicion d’une endocrinopathie et sa prise en charge nécessitent l’implication d’un spécialiste endocrinologue.
Le syndrome de Cushing (hypercorticisme endogène) est associé à une fréquence accrue de MASLD, avec des études radiologiques/biologiques rapportant de 20 à 34 % de stéatose chez ces patients, en particulier en présence d’obésité, de diabète et de dyslipidémie (23-25). La correction de l’hypercortisolisme s’accompagne habituellement d’une amélioration des anomalies hépatiques.
Une maladie de Wilson débutante peut être associée à une stéatose hépatique micro et/ou macro vacuolaire, au moins visible à la biopsie hépatique. Sa prévalence est de 1/30 000. Plus de 300 variants sont connus sur le gène ATP7B qui expliquent la maladie. La réalisation des marqueurs diagnostiques de la maladie de Wilson est utile à faire en cas de doute (cuprurie des 24 heures, céruléoplasminémie, rapport cuivre échangeable sur cuivre total). En cas de confirmation diagnostique, un traitement spécifique sera initié (5).
Une maladie cœliaque est à rechercher en cas de perturbations inexpliquées du bilan hépatique. De plus, une association entre une maladie cœliaque et une MASLD a été rapportée dans la littérature. Ainsi, une maladie cœliaque serait présente chez 3,4 à 7 % des adultes avec une MASLD. Les mécanismes expliquant cette association serait la fréquence, au cours de la maladie cœliaque, d’une pullulation bactérienne intestinale, d’une malabsorption des nutriments et d’un syndrome hyperperméabilité intestinal (5). En cas de confirmation diagnostique un régime sans gluten et un suivi spécifique seront initiés.
Stéatose hépatique et psoriasis
Une association entre MASLD, syndrome métabolique et psoriasis a été faite. Le risque serait plus grand chez les patients avec une atteinte articulaire. Le facteur commun entre la MASLD et le psoriasis serait l’inflammation systémique de bas grade. Le méthotrexate, qui est souvent utilisé au cours du psoriasis, n’aurait qu’un rôle mineur dans la survenue des éventuelles lésions hépatiques (5). .
L’exposition à certains polluants ou perturbateurs endocriniens pourrait aggraver une MASLD ou participer à une stéatose hépatique secondaire [substances per- et polyfluoroalkylées (PFAS), polychlorobiphényles (PCB), perchloroéthylène, chlorure de vinyle, dioxines, triflumizole, fludioxonil, tributylétain, bisphénol A (BPA), métaux lourds (plomb, arsenic, mercure, cadmium)] (5). Les stéatoses hépatiques secondaires liées à ces éléments toxiques sont nommées dans la littérature anglosaxonne toxicant-associated steatosis or steatohepatitis (TASH). Leur diagnostic formel est actuellement très difficile. La notion de profession exposée à ce type de substances peut orienter (26). Il n’existe pas de test utilisable en routine pour facilement diagnostiquer les personnes exposées. La fréquence et la sévérité des expositions à ces substances ne sont pas non plus clairement connues. Cela pourrait représenter un problème de santé publique qui concernerait l’ensemble de la population, y compris les femmes enceintes. Les mesures préventives incluant le respect des mesures de protection collectives et individuelles des travailleurs exposés, le choix de lieux de vie privée loin de zones industrielles contaminées et une alimentation d’origine biologique pourraient limiter les contaminations.
Certaines situations cliniques très particulières sont associées à une stéatose hépatique aiguë micro vésiculaire et présentent une gravité.
La stéatose hépatique aiguë gravidique (SHAG) est un diagnostic rare mais gravissime qui doit être évoqué de façon systématique lors de l’apparition, généralement brutale, d’une perturbation importante du bilan hépatique (hépatite aiguë sévère, voire hépatite fulminante), généralement associé à d’autres anomalies cliniques ou biologiques (troubles de la coagulation) peu spécifiques, en fin de troisième trimestre de grossesse. Le diagnostic peut être suspecté, par élimination, en l’absence des signes cardinaux faisant évoquer un foie toxémique ((pré)éclampsie ou syndrome HELLP). La stéatose est microvacuolaire et donc habituellement non visible à l’échographie. Une biopsie hépatique est rarement réalisée, compte tenu d’un rapport bénéfique-risque limite. En effet, la suspicion d’une SHAG nécessite un traitement qui est une grande urgence obstétricale. La patiente parturiente doit être transférée en maternité de niveau 3 et avoir, au plus tôt, un accouchement généralement par césarienne. Les anomalies hépatiques et extra hépatiques s’améliorent rapidement après la réalisation de l’accouchement, comme généralement dans les hépatopathies gravidiques (5).
Le syndrome de Reye favorisé par une infection et une prise d’aspirine à forte dose notamment chez l’enfant, est associé à une stéatose aiguë micro vésiculaire. Il est devenu très rare, grâce à la limitation de l’usage de l’aspirine à visée antipyrétique.
Lors de l’examen physique, rechercher une lipodystrophie
La présence d’une lipodystrophie peut être un élément-clef pour expliquer une stéatose hépatique. Les lipodystrophies sont classées selon leur présentation clinique (généralisée ou locale) et selon leur contexte de survenue (congénital ou acquis) (tableau 2) (27-31). Le plus souvent, l’hépato-gastro-entérologue, prenant en charge des adultes, sera confronté à une lipodystrophie généralisée acquise liée à une multithérapie du VIH (cf. paragraphe ci-dessus). Les autres causes sont beaucoup plus rares (prévalence 1/106) et sont liées principalement à une mutation d’un gène impliqué dans la croissance, la maturation ou la sénescence des cellules adipocytaires. Des causes auto-immunes/auto-inflammatoires peuvent également être présentes et se manifester par des panniculites (inflammation locale du tissu adipeux sous-cutané) puis une atrophie du tissu adipeux sous-cutané (27,28).
Causes
Âge de survenue
Traitement
Lipodystrophies congénitales
Lipodystrophie généralisée congénitale
Gène muté : AGPAT2, BSCL2, CAV1, CAVIN1.
Enfance
Mesures hygiéno-diététiques, fibrate, insulinothérapie si diabète, Metrleptine.
Mesures hygiéno-diététiques, fibrate, thiazolidinedione*, insulinothérapie si diabète, Metrleptine ?
Lipodystrophiesacquises
Lipodystrophie acquise généralisée
Exposition à une HAART du VIH.
Adulte
Mesures hygiéno-diététiques, fibrate, thiazolidinedione*, insulinothérapie si diabète. Modification du traitement HAART.
Maladie auto-immune.
Mesures hygiéno-diététiques, fibrate, insulinothérapie si diabète. Metrleptine Traitement de la maladie auto-immune. Minimalisation des corticostéroïdes.
Lipodystrophie acquise partielle
Maladie auto-immune.
Enfance
Mesures hygiéno-diététiques, fibrate, thiazolidinedione*, insulinothérapie si diabète, Traitement de la maladie auto-immune. Minimalisation des corticostéroïdes, Metrleptine ?
Tableau 2 : Principales lipodystrophies avec les causes, l’âge de survenue et les traitements recommandés, [adapté d’après (27,28,31)] : Abréviations et sigles : * : non disponible en France. ADRA2A : Adrenergic receptor, Alpha-2A subtype, AGPAT2 : 1-acyglycerol-3-phosphate O-acyltranferase, AKT2 : v-akt murine thymoma viral oncogene homolog, BSCL2 : Berardinelli-Seip congenital lipodystrophy, CAV1 : caveolin 1, CAVIN1 : Caveolae associated protein 1, CIDEC : cell death-inducing DFFA-like effector C, HAART : Highly Active Antiretroviral Therapy, LIPE : hormone sensitive lipase, LMNA : Lamin A/C, PCYT1A : Phosphate Cytidylyltransferase 1, Choline, Alpha, PLIN1 : perilipin 1, PPARG : peroxisome proliferator-activated receptor Gamma,
Beaucoup des lipodystrophies héréditaires se manifestent durant l’enfance. Certains adultes peuvent néanmoins avoir une présentation tardive. Une hypertriglycéridémie peut être présente au-delà de 5 g/L, avec parfois des épisodes antérieurs de pancréatite aiguës hypertriglycéridémiques.
Les associations entre ces maladies et les mutations génétiques présentes sont relativement bien connues (tableau 2). Compte tenu de la diminution du coût des tests génétiques, ceux-ci (génotypage ciblé, panels spécifiques ou séquençage « Whole Exome ») doivent être prescrits afin de diagnostiquer précisément la maladie présente.
Le diagnostic précis est en effet important car certaines lipodystrophies, en particulier les généralisées et certaines partielles, peuvent être traitées par la mise en place d’un analogue recombinant de la leptine (metreleptine). Ce traitement permet une amélioration des différentes complications dysmétaboliques. Les autres mesures comportent une diététique équilibrée, la prescription de metformine et d’hypolipémiant (fénofibrate, oméga 3) (27,28).
(re)Analyser le bilan lipidique sanguin
Un examen méthodique du bilan lipidique donné par « l’exploration d’une anomalie lipidique » doit être fait. Un niveau indosable de LDL cholestérol et un niveau effondré de triglycérides sont évocateurs d’une abétalipoprotéinémie. Un niveau très bas de LDL cholestérol et un niveau très bas de triglycérides sont évocateurs d’une hypobétalipoprotéinémie familiale biallélique. Un niveau légèrement abaissé de LDL cholestérol et de triglycérides peut être évocateur d’une hypobétalipoprotéinémie familiale mono allélique ou d’une maladie de rétention des chylomicrons. Dans la maladie de rétention des chylomicrons, une élévation de la CPK peut être observée. Tableau 3 (32,33).
Cause et fréquence
Principales anomalies
Risque évolutif
Traitement
Abétalipoprotéinémie
Gène muté : MTTP. Prévalence <1/106
Stéatose (100 %)
CHC
Régime pauvre en lipides, supplémentation en vitamines liposolubles
Hypobétalipoprotéinémiefamiliale
Gène muté : APOB, ANGPTL3, (PCSK9). Prévalence 1/1000 à 1/3000
Stéatose (50 %)
CHC
Régime pauvre en lipides, supplémentation en vitamines liposolubles
Déficit en LAL
Gène muté : LIPA. Prévalence 1/40 000 à 1/300 000 selon l’ethnie.
Stéatose micro vésiculaire, parfois micro+macro vacuolaire
Cirrhose, CHC
Régime très pauvre en lipides, enzyme de substitution : sébélipase alfa
Glycogénoses
Type I, III, IV, VI, IX, XI 1/25000 à 1/40000
Fibrose, cirrhose, CHC
Diététique, enzyme de substitution.
Intolérance héréditaire au fructose
Gène muté : ALDOB. 1,3 % des nouveau-nés aurait une copie mutée.
Stéatose macro vacuolaire
Cirrhose
Réduction du fructose alimentaire (<2 à 2,5 g/j), du sucrose et du sorbitol dans le régime. Limiter les fruits, remplacer le fructose par du glucose, du maltose, amidon. Supplémentation en vitamine C et acide folique.
Maladies du cycle de l’urée
Mutation liée au chromosome X. Déficit en ornithine transcarbamylase, Déficit en carbamoylphosphate synthetase, Intolérance à la protéine Lysinurique.
Gène muté : SLC25A13 Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD), prévalence 1/17 000 Forme de l’adulte : citrullinémie de type 2 (CTLN2), prévalence 1/100 000 à 1/200 000.
NICCD : Cholestase néonatale. CTLN2 : chez l’adulte, altérations psychiques, hyperammoniémie, stéatose hépatique macro et micro vésiculaire.
Fibrose hépatique, CHC
Tableau 3 : Principales maladies monogéniques (hors lipodystrophie) associées à une stéatose hépatique [adapté d’après (5)] : ALDOB : Aldolase B, ANGPTL3 : angiopoietin-like 3, APOB : Apolipoprotein B, CHC : carcinome hépatocellulaire, LIPA : Lipase A, MTTP : microsomal triglyceride transfer protein, PCSK9 : proprotein convertase subtilisin kexin 9, SLC25A13 : Solute Carrier Family 25 Member 13.
Dans le cas d’une abétalipoproténiémie et dans certaines formes d’hypobétalipoprotéinémie familiale, des signes cliniques associés à différentes atteintes d’organes peuvent orienter le diagnostic comme un retard de croissance au cours de l’enfance, une ataxie cérébelleuse, une rétinite pigmentaire, une neuropathie périphérique axonale, une acanthocytose, une dysfonction cardiaque, des signes liées à une malabsorption des lipides et à une carence en vitamine liposolubles A, D, E, K (33).
Les atteintes hépatiques peuvent être variées (stéatose, stéatohépatite) et parfois sévères avec la description de cirrhoses « cryptogéniques » et de carcinomes hépatocellulaires.
Dans la maladie de rétention des chylomicrons, une hépatomégalie et une stéatose sont rapportés chez 20 % des patients, sans cas de cirrhose décrite jusqu’à présent. L’endoscopie digestive retrouve une muqueuse entérocytaire blanche et des entérocytes chargés de lipides à l’examen anatomopathologique (33).
L’incidence de l’abétalipoprotéinémie est très faible, inférieure à 1 pour 106. L’incidence peut être plus fréquente chez certaines populations (juifs ashkénazes). La prévalence de l’hypobétalipoprotéinémie est de 1 sur 700 à 3 000.
Le diagnostic de certitude sera obtenu par le génotypage des gènes suspectés (génotypage ciblé, panel de gènes ou whole exome sequencing). La prise en charge thérapeutique consiste en régime pauvre en lipides, une supplémentation en vitamines liposolubles A, D, E, K (33).
Les perturbations du bilan lipidique au cours du rarissime déficit en lipase acide lysosomal (LAL-D) ne sont pas spécifiques. Une augmentation du LDL cholestérol (souvent> 1,6 g/L, parfois> 2 g/L), avec un HDL cholestérol bas, et des triglycérides élevés ou modérément élevés, une ApoB augmentée, une ApoA diminuée, dans un contexte de perturbation du bilan hépatique et d’hépatomégalie (généralement micro vésiculaire, donc avec une stéatose non visible en imagerie) avec une splénomégalie peut évoquer cette maladie. La forme classique est une maladie génétique autosomique récessive, néonatale, grave, appelée déficit en lipase acide lysosomal rapidement progressive. Sa prévalence est d’environ 0,3 à 1,1 cas par million chez les personnes issues d’ancêtres européens. Des rares formes atténuées, dite à début tardif, liées à des mutations particulières, peuvent être diagnostiquées dans l’enfance, à l’adolescence, voire chez des adultes. Ces manifestations sont très rares avec environ 3,5 à 6 cas par million chez des personnes issues d’ancêtres européens (24,34,35).
En cas de suspicion diagnostique, la collecte de sang capillaire sur buvard peut être réalisée afin de rechercher une baisse d’activité enzymatique de la lipase acide lysosomal. En cas de baisse de l’activité, un génotypage spécifique du gène LIPA sera réalisé. La mutation entraîne une accumulation de lipides dans les lysosomes et est associée à une stéatose microvésiculaire à la biopsie hépatique.
La reconnaissance de cette maladie dans sa forme rapidement progressive permet d’initier une enzymothérapie substitutive (sebelipase alfa) et un régime pauvre en lipides (34,35).
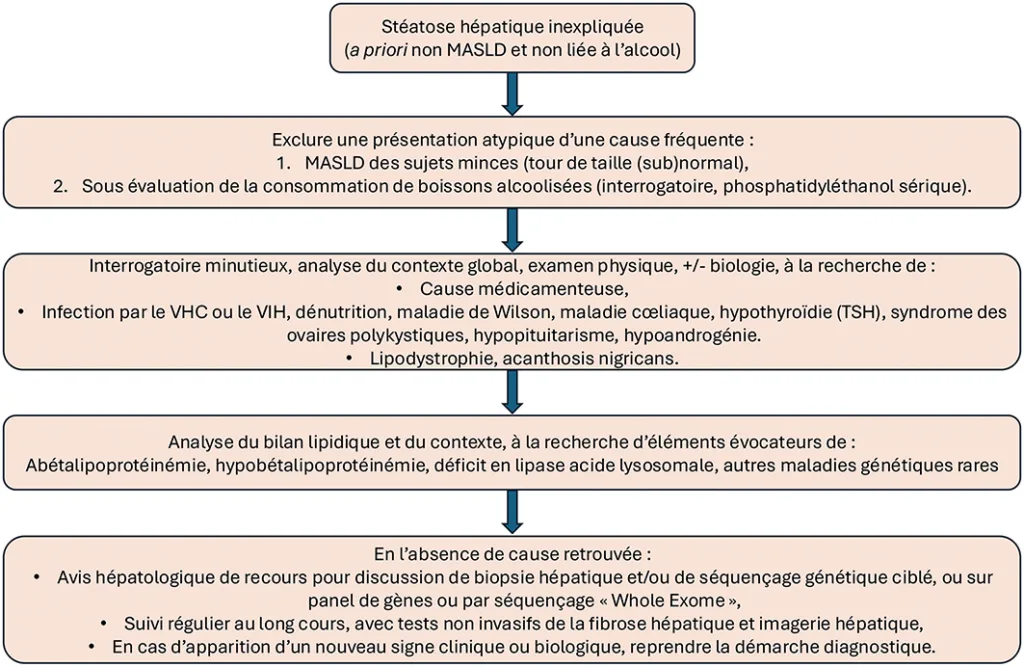
Figure 1 : Proposition d’algorithme diagnostique devant une stéatose hépatique inexpliquée
Que faire quand aucune cause n’est trouvée ?
Si à l’issue de l’enquête, aucune cause n’est retrouvée, il semble raisonnable d’évaluer la sévérité de la fibrose hépatique à l’aide de tests non invasifs et de proposer au patient un suivi tous les 1 à 2 ans. Une reprise de l’enquête étiologique pourra être faite dans le futur lors de l’apparition d’un nouveau signe clinique ou biologique ou lors de l’apparition d’une fibrose hépatique avancée. L’apparition d’une fibrose avancée devra faire rechercher et traiter d’éventuelles comorbidités hépatiques surajoutées.
Un avis spécialisé auprès d’un centre de recours hépatologique et/ou auprès d’un endocrinologue est pertinent. La réalisation d’une biopsie hépatique peut être proposée. Elle peut permettre d’orienter le diagnostic.
Il peut se discuter l’intérêt d’un séquençage génétique (ciblé ou sur un panel de gènes ou un séquençage « Whole Exome »). Cela peut permettre de trouver des mutations de gènes impliqués dans des maladies monogéniques du métabolisme les lipides ou autres (comme lors des lipodystrophies congénitales généralisées ou partielles, ou de l’a(hypo) béta lipoprotéinémie, d’un déficit en lipase acide lysosomal ou d’autres maladies métaboliques généralement révélées dans l’enfance), tableau 3 (5).
Cela peut également permettre de reconnaître la présence de polymorphismes génétiques fréquemment présents dans la population (en particulier celui de PNPLA3, mais également de TM6SF2, MBOAT7, HSD17B13) qui pourraient participer à la l’apparition d’une MASLD à composante principalement « génétique » (5).
La recherche de mutations liées à certaines maladies du foie bien connues pourrait également être intéressante. Ainsi, la mutation du gène HFE C282Y à l’état homozygote a été rapportée comme pouvant augmenter la MASLD d’un facteur 10. La co-présence des polymorphismes PNPLA3 et TM6SF2 augmenterait alors le risque de développement d’une cirrhose (5).
Les variants pathologiques dans le gène SERPINA1 peuvent conduire au déficit en alpha1 anti-trypsine caractérisé par un emphysème pulmonaire et/ou une maladie hépatique. La présence du variant c.1096G>A (p.E366K= p.E342K, rs28929474, MAF= 0,0184) qui encode la protéine Pi*Z est trouvée chez 2 à 4 % des Européens mais jusque 20 % des patients avec une MASLD. Les personnes homozygotes Pi*Z ont plus de stéatose et plus de fibrose hépatique sur les tests non invasifs, par rapport aux sujets normaux. L’hétérozygotie Pi*Z augmenterait le risque de progression vers une cirrhose MASH et l’apparition d’un CHC chez des patients présentant une MASLD (5).
Une forme particulière de MASLD familiale souvent accompagnée d’une dyslipidémie, à transition autosomique dominante, apparaissant après 40 ans, a été attachée à des mutations dans le gène ABHD5 qui code pour CGI-58 (comparative gene identification-58), qui est un co-facteur de ATGL (adipose triglyceride lipase). CGI-58 est impliqué dans la mécanique limitant l’hydrolyse des triglycérides en présence du variant pathologique PNPLA3 p.I148M (5).
En conclusion, en l’absence de cause dysmétabolique ou alcoolique, la recherche des causes rares de stéatose hépatique secondaire doit être faite. Certaines de ces causes rares peuvent coexister avec une MASLD, une MetALD ou une ALD. La démarche initiale est clinique basée sur un interrogatoire, un examen physique et l’analyse du bilan lipidique. Une biopsie hépatique, voire un séquençage génétique, pourront compléter l’enquête et confirmer ou infirmer une suspicion diagnostique. Plusieurs causes rares de stéatose hépatique secondaire ont un traitement spécifique. En cas de stéatose restant de cause inconnue, la poursuite d’une surveillance régulière de la fibrose hépatique par un/des test(s) non invasif(s) est probablement utile.
Références
Permutt Z, Le TA, Peterson MR, Seki E, Brenner DA, Sirlin C, et al. Correlation between liver histology and novel magnetic resonance imaging in adult patients with non-alcoholic fatty liver disease – MRI accurately quantifies hepatic steatosis in NAFLD. Aliment Pharmacol Ther 2012;36:22-29.
Caussy C, Alquiraish MH, Nguyen P, Hernandez C, Cepin S, Fortney LE, et al. Optimal threshold of controlled attenuation parameter with MRI-PDFF as the gold standard for the detection of hepatic steatosis. Hepatology 2018;67:1348-1359.
European Association for the Study of the Liver . Electronic address eee, European Association for the Study of D, European Association for the Study of O, European Association for the Study of the L. EASL-EASD-EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). J Hepatol 2024.
Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F, et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J Hepatol 2023;79:1542-1556.
Liebe R, Esposito I, Bock HH, Vom Dahl S, Stindt J, Baumann U, et al. Diagnosis and management of secondary causes of steatohepatitis. J Hepatol 2021;74:1455-1471.
Lee BP, Dodge JL, Terrault NA. National prevalence estimates for steatotic liver disease and subclassifications using consensus nomenclature. Hepatology 2024;79:666-673.
Fracanzani AL, Valenti L, Bugianesi E, Vanni E, Grieco A, Miele L, et al. Risk of non alcoholic steatohepatitis and fibrosis in patients with non alcoholic fatty liver disease and low visceral adiposity. J Hepatol 2010;11:11.
Golabi P, Bush H, Stepanova M, Locklear CT, Jacobson IM, Mishra A, et al. Liver Transplantation (LT) for Cryptogenic Cirrhosis (CC) and Nonalcoholic Steatohepatitis (NASH) Cirrhosis: Data from the Scientific Registry of Transplant Recipients (SRTR): 1994 to 2016. Medicine (Baltimore) 2018;97:e11518.
Long MT, Noureddin M, Lim JK. AGA Clinical Practice Update: Diagnosis and Management of Nonalcoholic Fatty Liver Disease in Lean Individuals: Expert Review. Gastroenterology 2022;163:764-774 e761.
Thurfjell A, Sandlund C, Adami J, Hasselstrom J, Hagstromer M, Lundh L. GPs’ experiences of phosphatidylethanol in treatment of hypertension: a qualitative study. BJGP Open 2023;7.
Dash A, Figler RA, Sanyal AJ, Wamhoff BR. Drug-induced steatohepatitis. Expert Opin Drug Metab Toxicol 2017;13:193-204.
Lopez-Pascual E, Rienda I, Perez-Rojas J, Rapisarda A, Garcia-Llorens G, Jover R, et al. Drug-Induced Fatty Liver Disease (DIFLD): A Comprehensive Analysis of Clinical, Biochemical, and Histopathological Data for Mechanisms Identification and Consistency with Current Adverse Outcome Pathways. Int J Mol Sci 2024;25.
Meunier L, Larrey D. Chemotherapy-associated steatohepatitis. Ann Hepatol 2020;19:597-601.
Di Martino V, Verhoeven DW, Verhoeven F, Aubin F, Avouac J, Vuitton L, et al. Busting the myth of methotrexate chronic hepatotoxicity. Nat Rev Rheumatol 2023;19:96-110.
Hosack T, Damry D, Biswas S. Drug-induced liver injury: a comprehensive review. Therap Adv Gastroenterol 2023;16:17562848231163410.
Gervasoni C, Cattaneo D, Filice C, Galli M, Gruppo Italiano Studio Nimi. Drug-induced liver steatosis in patients with HIV infection. Pharmacol Res 2019;145:104267.
Giralt M, Domingo P, Quesada-Lopez T, Cereijo R, Villarroya F. Lipodystrophy in HIV: Evolving Challenges and Unresolved Questions. Int J Mol Sci 2025;26.
Bessone F, Dirchwolf M, Rodil MA, Razori MV, Roma MG. Review article: drug-induced liver injury in the context of nonalcoholic fatty liver disease – a physiopathological and clinical integrated view. Aliment Pharmacol Ther 2018;48:892-913.
Zafirovska M, Zafirovski A, Rotovnik Kozjek N. Current Insights Regarding Intestinal Failure-Associated Liver Disease (IFALD): A Narrative Review. Nutrients 2023;15.
Addeo P, Cesaretti M, Anty R, Iannelli A. Liver transplantation for bariatric surgery-related liver failure: a systematic review of a rare condition. Surg Obes Relat Dis 2019;15:1394-1401.
Adams LA, Feldstein A, Lindor KD, Angulo P. Nonalcoholic fatty liver disease among patients with hypothalamic and pituitary dysfunction. Hepatology 2004;39:909-914.
Harrison SA, Bedossa P, Guy CD, Schattenberg JM, Loomba R, Taub R, et al. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. N Engl J Med 2024;390:497-509.
Rockall AG, Sohaib SA, Evans D, Kaltsas G, Isidori AM, Monson JP, et al. Hepatic steatosis in Cushing’s syndrome: a radiological assessment using computed tomography. Eur J Endocrinol 2003;149:543-548.
Chen K, Chen L, Dai J, Ye H. MAFLD in Patients with Cushing’s Disease Is Negatively Associated with Low Free Thyroxine Levels Rather than with Cortisol or TSH Levels. Int J Endocrinol 2023;2023:6637396.
Marengo M, Briet C, Munier M, Boursier J, Rodien P, Suteau V. Fatty Liver Disease Along Cushing Syndrome Evolution. J Clin Endocrinol Metab 2025;110:e2037-e2044.
European Association for the Study of the L, Clinical Practice Guideline Panel C, Panel m, representative EGB. EASL Clinical Practice Guideline: Occupational liver diseases. J Hepatol 2019;71:1022-1037.
Hussain I, Garg A. Lipodystrophy Syndromes. Endocrinol Metab Clin North Am 2016;45:783-797.
Patni N, Garg A. Lipodystrophy for the Diabetologist-What to Look For. Curr Diab Rep 2022;22:461-470.
Knebel B, Muller-Wieland D, Kotzka J. Lipodystrophies-Disorders of the Fatty Tissue. Int J Mol Sci 2020;21.
Zammouri J, Vatier C, Capel E, Auclair M, Storey-London C, Bismuth E, et al. Molecular and Cellular Bases of Lipodystrophy Syndromes. Front Endocrinol (Lausanne) 2021;12:803189.
Polyzos SA, Perakakis N, Mantzoros CS. Fatty liver in lipodystrophy: A review with a focus on therapeutic perspectives of adiponectin and/or leptin replacement. Metabolism 2019;96:66-82.
Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia. Curr Opin Lipidol 2014;25:161-168.
Bredefeld C, Hussain MM, Averna M, Black DD, Brin MF, Burnett JR, et al. Guidance for the diagnosis and treatment of hypolipidemia disorders. J Clin Lipidol 2022;16:797-812.
Balwani M, Balistreri W, D’Antiga L, Evans J, Ros E, Abel F, et al. Lysosomal acid lipase deficiency manifestations in children and adults: Baseline data from an international registry. Liver Int 2023;43:1537-1547.
de Las Heras J, Almohalla C, Blasco-Alonso J, Bourbon M, Couce ML, de Castro Lopez MJ, et al. Practical Recommendations for the Diagnosis and Management of Lysosomal Acid Lipase Deficiency with a Focus on Wolman Disease. Nutrients 2024;16.
FMC HGE : Organisme certifié Qualiopi pour la catégorie ACTIONS DE FORMATION.
Nous vous invitons à tester vos connaissances sur l’ensemble des QCU tirés des exposés des différents POST’U. Les textes, diaporamas ainsi que les réponses aux QCM seront mis en ligne à l’issue des prochaines journées JFHOD.