Connaître le bilan pré-thérapeutique et la classification anatomo-pathologique selon l’OMS
Connaître la prise en charge du syndrome carcinoïde
Connaître les indications du traitement chirurgical
Connaître les différents traitements systémiques et leurs indications
Les points forts
Les tumeurs neuroendocrines de l’intestin grêle (TNEG) ont une incidence d’environ 1,5/100 000. Leur classification anatomo-pathologique repose sur la différenciation cellulaire et le grade de l’OMS déterminé à partir de l’index de prolifération (Ki-67).
Le syndrome carcinoïde concerne environ 30 % des patients et associe classiquement diarrhée, flushs vasomoteurs et 5HIAA urinaire élevé. Sa principale complication au long cours est la cardiopathie carcinoïde. Son traitement repose sur les analogues de la somatostatine (SST).
Le bilan pré-thérapeutique inclut un scanner thoraco-abdominopelvien, une IRM hépatique avec séquences de diffusion, une TEP/TDM à la 18FDOPA et si une radiothérapie interne vectorisée (RIV) est envisagée, une TEP/TDM au 68Ga-DOTATOC.
La résection chirurgicale de la TNEG associée à un curage ganglionnaire mésentérique est le traitement de référence des formes non métastatiques. Elle peut être également envisagée dans les formes métastatiques en cas de lésion primitive symptomatique ou menaçante.
Pour les TNEG métastatiques non résécables, les analogues de la SST sont le traitement de référence de première ligne. En cas de progression et/ou de facteurs de mauvais pronostic, les principales options incluent la RIV, l’embolisation intra-artérielle hépatique, l’évérolimus et le cabozantinib. La chimiothérapie est généralement peu efficace.
5HIAA : acide 5-hydroxy-indolacétique CgA : chromogranine A
CNE : carcinome neuroendocrine
EIAH : embolisation intraartérielle hépatique G : grade tumoral
OMS : Organisation Mondiale de la Santé RIV : radiothérapie interne vectorisée SSP : survie sans progression
SST : somatostatine
TEP/TDM : tomographie par émission de positrons couplée au scanner TNE : tumeur(s) neuroendocrine(s)
TNEG : TNE de l’intestin grêle
Introduction
Les néoplasies neuroendocrines du tube digestif, auparavant appelées « tumeurs carcinoïdes », regroupent des tumeurs partageant des caractéristiques anatomo-pathologiques et pronostiques. Elles se divisent en tumeurs neuroendocrines (TNE) bien différenciées et carcinomes neuroendocrines (CNE) peu différenciés (1). Les TNE de l’intestin grêle (TNEG) proviennent de l’intestin moyen embryologique (« midgut ») et sont presque toujours bien différenciées, les carcinomes étant rarissimes dans cette localisation.
Les TNEG se développent à partir des cellules enterochromaffin-like du système endocrinien diffus situé dans la sous-muqueuse de l’intestin grêle. Elles sont caractérisées par leur aptitude à produire des peptides hormonaux dont la sérotonine, dont l’hypersécrétion peut aboutir à un syndrome carcinoïde. Plus de la moitié des TNEG est associée à des métastases. Les progrès récents des techniques d’imagerie, en particulier de médecine nucléaire, ont représenté une avancée notable pour la caractérisation ainsi que le bilan d’extension de ces tumeurs. Si la chirurgie demeure la pierre angulaire du traitement des TNEG, l’éventail thérapeutique s’est récemment enrichi de traitements systémiques ayant prouvé leur efficacité antitumorale dans les formes métastatiques, bien que la séquence thérapeutique optimale reste à définir. Chaque cas doit être discuté en RCP experte dans le cadre du réseau RENATEN. Cette revue vise à exposer les aspects diagnostiques, pronostiques et thérapeutiques des TNEG en s’appuyant sur les recommandations françaises et européennes les plus récentes (2-4).
Épidémiologie des TNEG
Les TNEG représentent moins de 1 % de toutes les tumeurs digestives et sont les formes les plus fréquentes des TNE du système digestif. Leur incidence est comprise entre 1,2 et 1,5 cas/100 000/an (5,6). En comparaison, l’incidence des TNE du pancréas et de l’appendice est d’environ 1 cas/100 000 an et celle des TNE de l’estomac et du rectum est d’environ 0,5 cas/100 000/an. L’incidence annuelle des TNEG a connu une augmentation constante au cours des dernières décennies, puisque celle-ci était d’environ 0,2-0,5 cas/100 000/an dans les années 1990 (5,6). Cette évolution résulte de divers facteurs, notamment de l’accroissement du nombre d’examens d’imagerie, de l’amélioration de la sensibilité des techniques de radiologie et de médecine nucléaire, d’une meilleure connaissance des TNE ainsi que d’une déclaration plus systématique dans les registres.
Le sex-ratio des TNEG est équilibré. Aucun facteur de risque spécifique n’a été déterminé à ce jour, bien que le risque de développer une TNEG serait augmenté en cas de tabagisme et/ou d’antécédents personnels/familiaux de cancers colorectal ou mammaire (7). La recherche d’une prédisposition génétique n’est actuellement pas recommandée pour les TNEG, mais les formes familiales existent, concerneraient 3-4 % des patients et semblent caractérisées notamment par l’existence de TNEG multiples.
Diagnostic anatomo-pathologique et classifications
Le diagnostic anatomo-pathologique de TNEG repose sur la différenciation morphologique neuroendocrine des cellules tumorales et l’expression d’au moins deux marqueurs immunohistochimiques (généralement, la chromogranine A [CgA], la synaptophysine et/ou INSM1) (1). La classification 2022 des néoplasies neuroendocrines selon l’OMS repose sur la différenciation histologique et le grade, évalué par l’index mitotique et/ou l’index Ki-67 (pourcentage de cellules tumorales marquées par l’anticorps MIB1 sur 2 000 cellules dans les hotspots) (1) (tableau 1). Ces éléments sont essentiels à l’évaluation pronostique des TNEG (8).
Différenciation
Index Ki-67
Index mitotique (mitoses pour 10 grands champs soit 2mm²)
Les « tumeurs neuroendocrines » sont par définition bien différenciées et sont composées de cellules monomorphes, de taille moyenne et à cytoplasme abondant. Leur noyau est généralement ovoïde et contient une chromatine « poivre et sel » et de petits nucléoles bien visibles. Ces cellules bien limitées sont généralement organisées en travées ou en foyers, avec peu de figures mitotiques, et entourées d’un stroma hypervascularisé. En règle générale, les TNEG sont le plus souvent G1, et seules environ 20 % d’entre elles ont un index Ki-67> 10 % ; les formes G3 sont très rares (5 % environ) et plus agressives (8,9). À l’inverse, les « carcinomes neuroendocrines » (CNE) sont par définition peu différenciées et peuvent être à petites ou à grandes cellules, présentent de nombreuses atypies cyto-nucléaires, des plages de nécrose et de nombreuses figures mitotiques. Ces formes, particulièrement agressives, sont rarissimes dans l’intestin grêle.
Les TNEG doivent également être classées selon leur stade Tumor-Nodes-Metastases (TNM) (tableau 2). Cette classification présente un intérêt pronostique majeur et permet une homogénéisation de la prise de décision thérapeutique.
TX
Tumeur non évaluable
T0
Pas de tumeur identifiable
T1
Envahit la lamina propria ou la sous-muqueuse et taille≤ 1 cm
Masse mésentérique> 2 cm et/ou> 12 ganglions métastatiques
Mx
Statut non évaluable
M0
Absence de métastase à distance
M1a
Métastases hépatiques
M1b
Métastases extra-hépatiques
M1c
Métastases hépatiques et extra-hépatiques
Tableau 2 : Classification TNM des TNEG selon la 8e édition de l’UICC (2017)
Manifestations cliniques des TNEG
Symptômes liés aux localisations tumorales
Le diagnostic des TNEG est généralement fortuit car la plupart d’entre elles sont de petite taille, de croissance lente et initialement asymptomatiques, ce qui explique un retard diagnostique de 5 ans en moyenne (10). Les symptômes liés aux tumeurs primitives sont souvent aspécifiques comme des troubles du transit, des douleurs abdominales (40 %), voire un syndrome subocclusif (20 %) ou plus rarement (5 %) un saignement digestif (9). Cela explique en partie le taux élevé (environ 50 %) de patients ayant des métastases à distance au moment du diagnostic initial. Ces métastases synchrones concernent le plus souvent le foie (75-85 %), les ganglions à distance (30-40 %), le péritoine (20-25 %) et/ou les os (10-15 %), et peuvent être symptomatiques par leur volume (douleurs) et/ou leur localisation (par exemple, obstruction digestive) (8,9).
Syndrome carcinoïde
Environ 25-40 % des TNEG métastatiques sont dites fonctionnelles, c’est-à-dire associées à un syndrome carcinoïde (3). Le syndrome carcinoïde est l’ensemble des symptômes liés à l’hypersécrétion de sérotonine principalement, ainsi que d‘histamine, dopamine, prostaglandines et tachykinines. Excrêtée dans la circulation portale par la TNEG primitive, la dégradation hépatique de la sérotonine entraine son élimination sanguine puis urinaire sous la forme d’acide 5-hydroxy-indolacétique (5HIAA). Le syndrome carcinoïde survient donc presque exclusivement si des métastases hépatiques sécrètent ces substances vasoactives directement dans la circulation systémique.
Les flushs (80-90 %) et la diarrhée (70-80 %) sont les deux principaux signes cliniques associés au syndrome carcinoïde. Les flushs sont des bouffées vasomotrices provoquées par divers facteurs tels que l’émotion, l’effort physique, l’alimentation ou la consommation d’alcool ; parfois spontanées. Leur durée est généralement de quelques minutes, mais ils sont parfois plus prolongés, voire peuvent devenir permanents. Ils se présentent sous forme d’érythème, voire de coloration violacée, avec un aspect en « carte de géographie » alternant zones rouges et blanches. Certains flushs peuvent provoquer un prurit et apparaissent principalement sur le visage, le cou et le tronc ; ils peuvent s’accompagner d’une vasodilatation conjonctivale et d’un larmoiement, mais sans hypersudation associée. Les patients présentant des flushs chroniques développent fréquemment des télangiectasies faciales (25 %) similaires à celles observées dans la rosacée. Enfin, ces flushs peuvent s’accompagner, de manière simultanée ou pas, d’un bronchospasme (15 %).
La diarrhée est surtout motrice (avec une part sécrétoire), impérieuse et généralement postprandiale, volontiers associée à un inconfort ou des douleurs abdominales. Toute diarrhée survenant dans l’évolution d’une TNEG n’est pas forcément liée à un syndrome carcinoïde, mais peut être causée par une pullulation microbienne en cas d’obstacle chronique de l’intestin grêle ou peut être la conséquence d’une résection iléocolique droite.
Une cardiopathie carcinoïde survient chez 30 % à 50 % des patients atteints de syndrome carcinoïde, et marque souvent un tournant péjoratif dans l’évolution de la maladie (3). Elle s’explique par une fibrose destructrice de l’endocarde et des valves cardiaques, essentiellement du cœur droit, consécutive à une exposition prolongée à la sérotonine sécrétée par les métastases hépatiques. La cardiopathie carcinoïde se manifeste donc le plus souvent par une insuffisance (ou régurgitation) tricuspidienne et/ou une sténose de la valve pulmonaire. À un stade plus tardif, un dysfonctionnement ventriculaire droit peut apparaître, et parfois une atteinte des valves gauches (notamment en cas de foramen ovale perméable et/ou de métastases pulmonaires). La cardiopathie carcinoïde peut parfois se développer alors que le syndrome carcinoïde peut être modéré, voire infraclinique mais prolongé. Ainsi, il est recommandé de réaliser une échographie cardiaque par un cardiologue spécialisé dans la recherche de la cardiopathie carcinoïde, tous les 2 ans chez tout patient atteint de TNEG métastatique, ou tous les ans en cas de syndrome carcinoïde et/ou de taux élevé de 5HIAA urinaire (2,3).
La crise carcinoïde correspond à l’exacerbation aiguë du syndrome carcinoïde. Elle peut entraîner des flushs sévères, une bronchoconstriction, des troubles du comportement, des troubles du rythme cardiaque et des changements importants de la pression artérielle pouvant aller jusqu’au collapsus cardiovasculaire. La crise carcinoïde est causée par un relargage aigu et massif de peptides hormonaux vasoactifs par la tumeur, le plus souvent provoquée par un traitement des lésions tumorales, en particulier les traitements locorégionaux notamment des métastases hépatiques, et est favorisée par toute anesthésie générale. La prévention repose sur l’administration péri-opératoire d’octréotide et d’antihistaminiques, et la non-utilisation des médicaments favorisant un relargage de catécholamines.
Manifestations biologiques des TNEG
La CgA est le marqueur plasmatique général des TNE. Néanmoins, les variations inter- et intra-individuelles sont importantes et les faux positifs sont fréquents, notamment en cas de prise d’inhibiteurs de la pompe à protons, de gastrite de toute cause (Infection à H.Pylori, maladie de Biermer) et/ou d’insuffisance rénale sévère (11). Un taux élevé de CgA devrait être systématiquement validé par une gastrinémie et une créatininémie normales. Le taux de CgA est corrélé au volume tumoral ; un taux élevé au diagnostic initial est donc un facteur pronostique péjoratif. Ainsi, sa sensibilité diagnostique est faible (10 %-40 %) pour les TNEG localisées contre 70 %-100 % au stade métastatique (11). Sa sensibilité est également plus élevée en cas de TNEG fonctionnelle. Le dosage répété de la CgA au cours du suivi n’a pas démontré son utilité en comparaison à l’imagerie, mais l’évolution du taux de CgA pouvant refléter celle de la masse tumorale, une élévation semble prédire une rechute ou une progression.
Le 5HIAA est le produit d’élimination de la sérotonine (dont le dosage plasmatique n’a pas d’intérêt en routine) et est donc spécifique des TNEG en comparaison aux autres localisations de TNE. La sensibilité et la spécificité du 5HIAA urinaire (> 50 µmol/24 h) pour le diagnostic de TNEG sont respectivement d’environ 70 % et 90 % (3,12). Ses performances sont meilleures en présence de métastases et/ou d’un syndrome carcinoïde. Des taux élevés de 5HIAA (à partir de 220 µmol/24 h) sont associés à un pronostic péjoratif et à la survenue d’une cardiopathie carcinoïde. Néanmoins, ce marqueur présente une variabilité inter- et intra-individuelle élevée et de nombreux faux positifs existent, notamment alimentaires. Pour minimiser ces biais, son recueil est réalisé sur 3 jours et doit être précédé d’un régime contraignant pauvre en tryptophane et sérotonine (2).
Bilan d’extension
Imagerie conventionnelle
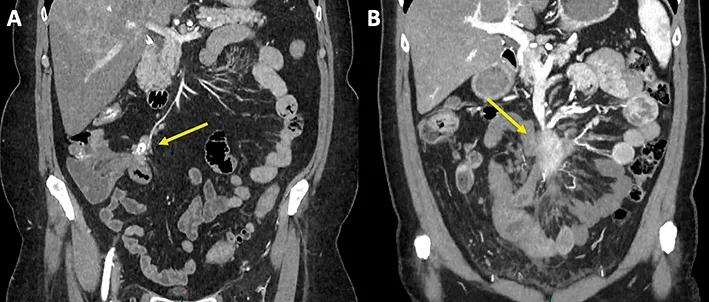
Le scanner et l’IRM sont utilisés en routine pour le diagnostic, le bilan d’extension et le suivi des TNEG (2). Les TNEG étant hypervascularisées, le scanner thoraco-abdominopelvien doit être réalisé avec acquisitions aux temps artériel tardif puis portal. Les TNEG sont fréquemment associées à des métastases ganglionnaires (souvent calcifiées) localisées le long des vaisseaux mésentériques avec mésentérite rétractile en relation avec une fibrose causée par la sécrétion de sérotonine, réalisant un « complexe mésentérique » (figure 1). Une ischémie veineuse grêlique peut être associée.
Figure 1 : Aspect scanographique des métastases ganglionnaires mésentériques associées aux TNEG : Coupes coronales de scanners abdominopelviens avec injection précoce de produit de contraste artériel. (A) amas ganglionnaire de petite taille, calcifié, avec minime rétraction mésentérique adjacente, le long de l’artère iléo-cæco-appendiculaire et loin du tronc de l’artère mésentérique, donc résécable à visée curative. (B) volumineux complexe ganglionnaire engainant le tronc des vaisseaux mésentériques supérieurs et les premières branches jéjunales, associée à une mésentérite rétractile compliquée d’une ischémie veineuse de quelques branches iléales, donc non résécable à visée curative.
Le scanner est également incontournable pour évaluer la présence de métastases, notamment hépatiques qui sont le plus souvent hyperdenses à la phase artérielle et hypodenses à la phase portale (13). L’envahissement tumoral hépatique peut être évalué de manière reproductible selon une échelle semi-quantitative (0-10 %, 11-25 %, 26-50 % et >50 %).
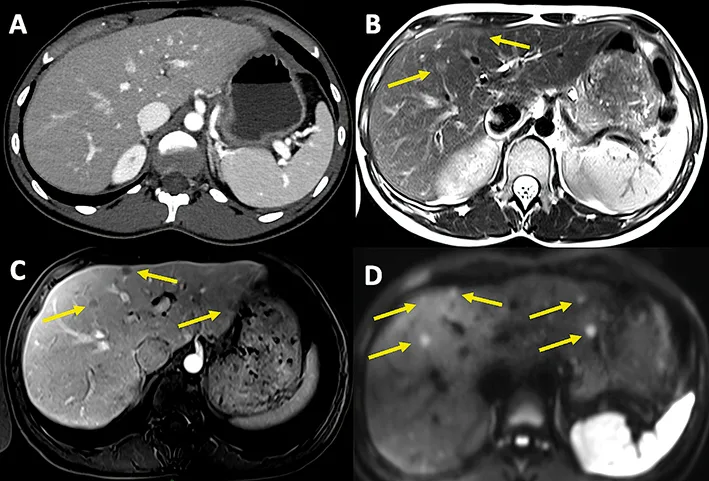
L’IRM est indispensable au bilan d’extension hépatique, en particulier lorsque les métastases paraissent résécables (2,4). En effet, elle est plus performante que le scanner et l’imagerie des récepteurs de la somatostatine pour la détection et la caractérisation des métastases hépatiques. Les séquences les plus performantes sont les séquences de diffusion, la séquence T1 avec injection de gadolinium et acquisition au temps artériel et la séquence T2-FSE (Figure 2) (14).
Figure 2 : Scanner et IRM hépatique dans le cadre du bilan d’extension d’une TNEG : (A) Scanner avec injection de contraste au temps artériel, et IRM pondérée en T2 (B), en T1 avec injection de gadolinium au temps artériel (C) et en séquence de diffusion (D), montrant – dans cet ordre – un nombre croissant de métastases hépatiques bilobaires.
Imagerie nucléaire
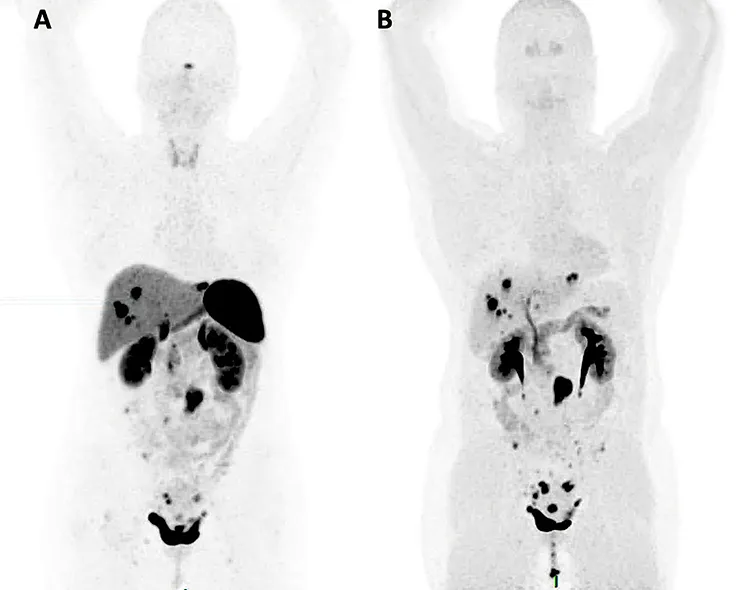
La quasi-totalité des TNEG expriment des récepteurs de la somatostatine (SST), qui sont visualisables après injection intraveineuse d’un analogue de la SST couplé à un isotope radioactif. La tomographie par émission de positrons (TEP) couplée au scanner (TDM) utilisant un analogue de la SST marqué au 68Gallium (en France, le 68Ga-DOTATOC) permet donc de réaliser une cartographie « corps entier » des récepteurs à la SST. La TEP/TDM au 68Ga-DOTATOC possède une sensibilité d’environ 90 % pour la détection des TNEG et des métastases associées (15), ce qui est bien supérieur à la sensibilité de la scintigraphie à l’111Indium-pentetreotide (Octréoscan®) (70 % environ) qui était utilisée auparavant (figure 3A). Elle est donc recommandée dans le bilan d’extension de toute TNEG (2,4). Une expression forte des récepteurs à la SST est associée à un pronostic plus favorable et prédit une bonne efficacité de la radiothérapie interne vectorisée (RIV) notamment lorsque la fixation tumorale est supérieure à celle du foie non tumoral. La spécificité de la TEP/TDM au 68Ga-DOTATOC est supérieure à 90 %. Les quelques faux positifs incluent les méningiomes, mélanomes, cystadénomes séreux, les granulomatoses (sarcoïdose, tuberculose) et les rates accessoires.
La TEP/TDM à la 18fluoro-dihydroxy-phénylalanine (18FDOPA) mesure l’activité des transporteurs des acides aminés et des enzymes décarboxylases, qui est élevée (et spécifique) dans les TNEG. Ses performances diagnostiques pour la détection des lésions primitives et métastatiques de TNEG sont très élevées (90-95 %), et supérieures à celles de l’imagerie morphologique (figure 3B). De plus, la TEP/TDM à la 18FDOPA a une sensibilité plus élevée que la TEP/TDM au 68Ga-DOTATOC à l’échelle lésionnelle (16). La TEP/TDM à la 18FDOPA ne donne pas d’information théranostique mais sa précision la rend très utile en vue de réaliser un bilan d’extension précis, notamment avant une intervention thérapeutique lourde – en particulier chirurgicale (2).
La TEP/TDM au 18fluoro-déoxyglucose (18FDG) est spécifique du métabolisme de toute maladie tumorale agressive, et apporte surtout une information pronostique. Elle représente donc l’imagerie nucléaire de référence pour les carcinomes neuroendocrines peu différenciés (dont les formes grêliques sont rarissimes). La TEP/TDM au 18FDG peut également être utile chez les patients ayant une TNEG sans fixation en TEP/TDM au 68Ga-DOTATOC, qui sont rares mais peuvent s’observer en cas d’index de prolifération élevé (G3 généralement).
Figure 3 : Examens d’imagerie nucléaire du bilan d’extension des TNEG : TEP/TDM au 68Ga-DOTATOC (A) et TEP/TDM à la 18FDOPA (B) chez une patiente de 51 ans ayant une TNEG classée G2 (Ki-67 7 %), associée à des métastases ganglionnaires mésentériques, hépatiques, péritonéales et ovariennes. Les deux examens montrent un nombre similaire d’organes métastatiques mais la TEP/TDM à la 18FDOPA montre un plus grand nombre de lésions, notamment grâce à un bruit de fond moins important en particulier au niveau hépatique.
Principaux facteurs pronostiques des TNEG
Les principaux facteurs pronostiques des TNEG incluent (8,9,17) :
La classification histologique selon l’OMS décrite ci-dessus (tableau 1), selon le degré de différenciation et l’index de prolifération, qui sont des facteurs pronostiques majeurs.
Le stade tumoral selon la classification TNM (Tableau 2). Environ 10-20 %, 30-40 % et 40-60 % des TNEG sont diagnostiquées respectivement à un stade localisé, localement avancé et métastatique. Les taux de survie à 5 ans respectifs sont 90-100 %, 70-90 % et 60-75 %.
Le volume métastatique, évaluable par le pourcentage d’envahissement hépatique, l’existence de métastases extra-hépatiques (en particulier une carcinose péritonéale et/ou des métastases osseuses) et le nombre de sites métastatiques.
De manière concordante, des taux élevés de marqueurs tumoraux (CgA plasmatique, 5HIAA urinaire) sont associés à un plus mauvais pronostic.
L’existence d’un syndrome carcinoïde est associée à un pronostic défavorable, en particulier lorsqu’il se complique d’une cardiopathie carcinoïde, qui est directement responsable ou associée à environ 20 % des décès en lien avec les TNEG.
La pente évolutive, c’est-à-dire la rapidité de progression morphologique de la maladie, spontanément ou sous traitement.
Enfin, le sexe masculin, l’âge avancé, la présence de symptômes, une fixation faible en TEP/TDM au 68Ga-DOTATOC et/ou une fixation intense en TEP/TDM au 18FDG sont des facteurs de mauvais pronostic, bien que peu confirmés prospectivement.
Traitement du syndrome carcinoïde
Le traitement des symptômes liés au syndrome carcinoïde est prioritaire et doit être débuté dès le diagnostic suspecté (2-4). Ce traitement repose principalement sur les analogues de la SST, qui sont efficaces à visée antisécrétoire. Il existe deux analogues de la SST à libération prolongée (LP) : l’octréotide (une injection intramusculaire de 10, 20 ou le plus souvent 30 mg tous les 28 jours) et le lanréotide (une injection sous-cutanée profonde de 60, 90 ou le plus souvent 120 mg tous les 28 jours) (3,18). Ces deux traitements permettent d’obtenir de manière similaire un soulagement rapide de la diarrhée (60-70 %), des flushs (70-80 %) et une réduction des marqueurs biochimiques (40-50 %). Les effets secondaires sont bénins, incluant principalement une insuffisance pancréatique exocrine (30-40 %), un déséquilibre du diabète et de possibles complications lithiasiques biliaires. L’existence d’une souffrance ischémique intestinale (généralement secondaire à une mésentérite rétractile sévère), est la principale situation de précaution voire de restriction d’utilisation des analogues de la SST car ceux-ci peuvent parfois aggraver l’ischémie digestive en raison d’une diminution de la perfusion viscérale.
En cas de syndrome carcinoïde sévère, l’octréotide à libération immédiate (50, 100 ou 200 μg) est utilisé pour contrôler rapidement les symptômes. Le syndrome carcinoïde peut être ou devenir réfractaire aux analogues de la SST chez environ 30 % des patients. Après avoir éliminé un problème d’observance, d’autres causes de diarrhée doivent être écartées, notamment une pullulation microbienne en cas d’obstacle chronique intestinal, une conséquence mécanique et/ou fonctionnelle d’une résection iléocolique droite, et une diarrhée liée à l’insuffisance pancréatique exocrine secondaire aux analogues de la SST. Les analogues de la SST peuvent alors être administrés à doses augmentées et/ou rapprochées (3,18). Le télotristat est un inhibiteur de la tryptophane hydroxylase, diminue la synthèse périphérique de la sérotonine et peut entraîner une amélioration de la diarrhée et une diminution du taux urinaire de 5HIAA, sans toutefois diminuer le nombre de flushs. Cependant, il n’est pas disponible en France. Enfin, l’interféron alpha peut également être utilisé en cas de syndrome carcinoïde réfractaire.
Traitement de la tumeur primitive et des adénopathies régionales
Le traitement de référence des TNEG non métastatiques est la résection chirurgicale de la tumeur primitive (multiples dans 20-30 % des cas) associé à un curage mésentérique (2,4). Cette résection, à visée curative lorsque toute la maladie peut être réséquée, doit préserver au maximum la longueur de l’intestin grêle, voire quand cela est possible la valvule iléo-cæcale afin de limiter les séquelles de la chirurgie.
Toute la cavité abdominale doit être explorée à la recherche de métastases à distance, en particulier le parenchyme hépatique et le péritoine avec exploration des ovaires. La palpation manuelle de tout l’intestin grêle pour détecter les TNEG multiples (20-30 %) est un temps indispensable de l’intervention. Un curage ganglionnaire mésentérique doit être systématiquement effectué quelle que soit la taille de la tumeur primitive car des métastases ganglionnaires sont présentes dans plus de 80 % des cas (2,19). Le curage doit permettre l’analyse d’au moins 8 ganglions lymphatiques pour améliorer le pronostic et pour correctement identifier les tumeurs N+. Un angioscanner abdomino-pelvien de qualité permettant de visualiser les rapports entre la masse mésentérique et les vaisseaux mésentériques doit être réalisé en préopératoire pour prédire les difficultés de résection de la masse mésentérique (19). Le curage mésentérique est faisable si au moins 3 branches artérielles à visée jéjunales sont libres d’envahissement tumoral. Une telle intervention doit être réalisée en centre expert.
En cas de complication aiguë en rapport avec une TNEG ou une masse mésentérique nécessitant une intervention chirurgicale d’urgence, le geste chirurgical doit se concentrer sur le problème immédiat (occlusion intestinale, ischémie mésentérique). Afin d’éviter une ré-opération ultérieure difficile, il convient de réaliser une résection limitée de la zone intestinale malade, avec ou sans rétablissement de la continuité et sans dissection intrapéritonéale étendue. Un bilan d’extension précis en vue d’une potentielle réintervention carcinologique à froid sera réalisé en postopératoire. En cas de maladie métastatique, la chirurgie des TNEG et des lésions mésentériques associées doit être systématiquement envisagée en cas de symptômes en rapport avec ces lésions (occlusion, ischémie mésentérique, douleur). En l’absence de symptômes et si la masse mésentérique est résécable, la résection chirurgicale doit également être discutée en RCP experte selon l’état général du patient et le projet thérapeutique (2). L’impact de cette stratégie sur la survie reste débattu bien qu’un effet bénéfique ait été rapporté. La résection ayant notamment pour but de limiter le risque de chirurgie en urgence, elle paraît donc particulièrement justifiable en cas masse mésentérique jugée « menaçante ».
Une cholécystectomie doit être systématiquement associée en cas de chirurgie digestive chez les patients atteints de TNEG métastatique ou à risque de récidive métastatique, en raison des risques ultérieurs de lithiase vésiculaire associé aux analogues de la SST et de cholécystite ischémique liée à l’embolisation intra-artérielle hépatique.
Traitement de la maladie métastatique
Stratégie globale de prise en charge
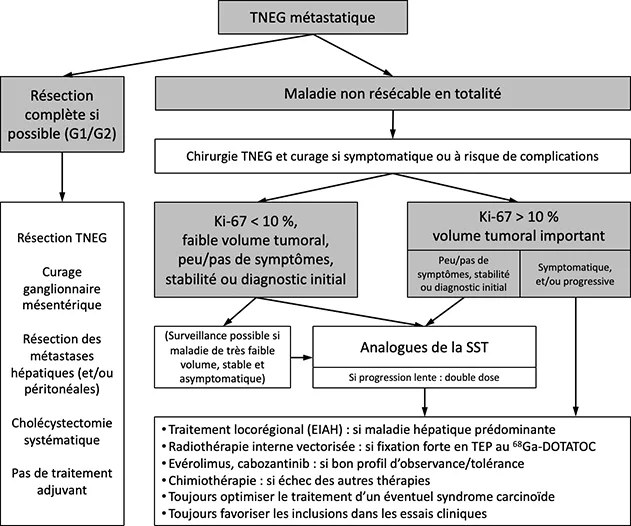
Les patients ayant une TNEG métastatique peuvent avoir une survie très prolongée (taux de survie globale à 5 ans d’environ 70 %). Du fait de la rareté de cette situation, le nombre d’essais randomisés et donc le niveau de preuves scientifiques est globalement faible. Les différents traitements disponibles ont été peu ou pas comparés entre eux prospectivement, et les séquences thérapeutiques optimales ne sont pas bien définies. Du fait de la survie généralement prolongée des patients, la stratégie doit chercher à éviter une toxicité séquellaire des traitements, qui risque d’altérer la qualité de vie et d’empêcher des traitements ultérieurs. Les décisions thérapeutiques reposent majoritairement sur des critères pronostiques évalués individuellement plutôt que sur des critères prédictifs d’efficacité. Tous les dossiers de TNEG doivent donc être discutés au cas par cas dans le cadre de RCP expertes du réseau RENATEN (https://www.reseau-gte.org/renaten/). La figure 4 propose un algorithme de traitement adapté des recommandations françaises et européennes (2,4).
Figure 4 : Proposition d’algorithme de traitement des patients atteints de TNG métastatique
Traitement chirurgical des métastases
La résection et/ou la destruction (percutanée ou peropératoire, par micro-ondes ou radiofréquence) de toutes les métastases visibles doit toujours être discutée, éventuellement en combinant différentes modalités, lorsque celle-ci semble réalisable chez un patient n’ayant pas de métastases extra-hépatiques prédominantes et une croissance tumorale lente (2,20). Le taux de survie sans récidive à 5 ans est de 91 % en cas de résection complète des métastases hépatiques, contre 76 % en cas de résection incomplète. Bien que la résécabilité soit compromise en cas d’envahissement hépatique> 50 %, l’atteinte bilobaire n’est pas une contre-indication. Dans cette situation, une chirurgie en deux temps peut être envisagée en centre expert et permet d’obtenir des taux de survie globale d’environ 90 % (2,20). La morbidité et la mortalité des procédures extensives et le volume hépatique restant anticipé doivent être mises en balance avec le risque presque inévitable de récidive métastatique à long terme.
En cas de carcinose péritonéale diffuse, une résection chirurgicale initialement prévue ne doit pas être réalisée (sauf en situation d’urgence), afin de pouvoir conserver la possibilité ultérieure d’une chirurgie péritonéale en centre expert. La cytoréduction chirurgicale complète de la carcinose péritonéale doit être envisagée lorsque celle-ci paraît possible, car elle semble être associée à un bénéfice de survie (21). L’ajout d’une chimiothérapie intrapéritonéale hyperthermique est en revanche associée à une morbidité bien plus élevée sans bénéfice de survie, et n’est pas recommandé.
Une transplantation hépatique peut être discutée en cas de métastases hépatiques diffuses non résécables, non ou très lentement évolutives, avec un Ki-67 bas (< 10 %, non déterminé précisément), en l’absence de métastases extra-hépatiques, chez des patients jeunes (< 60 ans), sans hépatomégalie initiale (et avec un envahissement hépatique< 50 %), chez qui la TNEG a été réséquée et après un recul évolutif suffisant à partir du diagnostic de la maladie métastatique (2).
Analogues de la somatostatine à visée antitumorale
Les analogues de la SST ont une action antiproliférative sur les TNEG par des mécanismes directs sur la signalisation et le métabolisme cellulaire, et indirects sur le microenvironnement tumoral [18]. L’étude de phase III PROMID a démontré que l’octréotide LP prolongeait la survie sans progression (SSP) en comparaison à un placebo chez des patients atteints de TNEG avancées de bas grade avec faible envahissement hépatique (médiane 14,3 mois versus 6 mois, p< 001) (22). L’étude de phase III CLARINET a confirmé ces résultats avec le lanréotide, en étendant la population cible aux TNE avec Ki-67< 10 % et/ou à envahissement hépatique< 50 % (médiane 32,8 mois, vs. 18 mois dans le groupe placebo, p< 001) (23). Ces études ont permis de positionner les analogues de la SST comme le traitement de première ligne en cas de TNEG métastatique sans facteur pronostique péjoratif (progression faible ou nulle, volume tumoral faible, Ki-67 faible, symptômes absents ou contrôlés). Alternativement, une surveillance active peut être envisagée en cas de maladie minime ou non-mesurable et si le risque évolutif est évalué comme faible.
En cas de stabilisation prolongée sous analogues de la SST à dose standard en première ligne, leur dose peut être augmentée, en rapprochant l’intervalle des injections à 14 jours, sans toxicité exacerbée, notamment en cas d’index Ki-67< 10 % (24). Cette stratégie permet de retarder l’utilisation des autres traitements – certes plus efficaces mais également plus toxiques, élément important chez les patients atteints de TNEG métastatique dont la survie est généralement prolongée.
Embolisation intra-artérielle hépatique
Les métastases hépatiques des TNEG ont une vascularisation riche et essentiellement artérielle, alors que le foie non tumoral est essentiellement vascularisé par la veine porte. Ainsi, l’embolisation intra-artérielle hépatique (EIAH) peut induire une ischémie tumorale tout en préservant le foie sain. L’EIAH est recommandée si la maladie hépatique est prédominante, en cas de progression tumorale ou d’emblée en cas de facteurs pronostiques péjoratifs comme des symptômes spécifiques, et/ou un syndrome carcinoïde difficile à contrôler (2,4,25). L’EIAH permet d’obtenir une réponse tumorale dans environ 40 % des cas avec une SSP médiane comprise entre 12 et 18 mois ; plusieurs traitements successifs pouvant être réalisés.
Les contre-indications à l’EIAH sont l’anastomose ou l’endoprothèse biliaire, la dilatation des voies biliaires et la thrombose portale complète. À l’inverse, elle n’est pas strictement contre-indiquée en présence de métastases extra-hépatiques non prédominantes, non progressives et asymptomatiques. Pour réduire la morbidité, le volume du foie doit être segmenté en 2-3 zones à traiter successivement avec un délai de 4 à 8 semaines entre chacune. La chimio-embolisation ne semblant pas plus efficace que l’EIAH seule pour les métastases de TNEG, cette dernière peut donc être préférée dans ce contexte (25). La radiothérapie intra-artérielle hépatique sélective à l’Yttrium 90 n’est pas disponible en routine en France et n’est actuellement pas remboursée dans l’indication des TNEG. Néanmoins, les données d’études rétrospectives suggèrent une efficacité au moins équivalente à celle de l’embolisation et possiblement une meilleure tolérance à court terme. Elle pourrait être intéressante en cas de métastases hépatiques regroupées en masse limitée à une partie du foie (2,25).
Radiothérapie interne vectorisée (RIV) au 177Lu-DOTATATE
La RIV consiste en l’injection intraveineuse d’un peptide couplé à une particule radioactive et capable de reconnaître une cible spécifique. Le premier traitement de RIV autorisé et remboursé dans les TNEG est le 177Lu-DOTATATE, qui consiste en un analogue de l’octréotide radiomarqué au 177Lutétium. La fixation du 177Lu-DOTATATE sur les récepteurs de la SST et l’internalisation dans les cellules tumorales entraîne leur destruction par les particules radioactives. L’utilisation du 177Lu-DOTATATE ne peut s’envisager qu’en cas d’expression forte des récepteurs de la SST, correspondant à une fixation des localisations tumorales supérieure à celle du foie non tumoral en TEP/TDM au 68Ga-DOTATOC. Bien que la tolérance soit généralement excellente, la RIV peut entraîner une toxicité à long terme sur la moelle osseuse avec un risque d’hématotoxicité, et un risque faible mais réel d’hémopathie maligne secondaire.
L’étude de phase III NETTER-1 a démontré que le 177Lu-DOTATATE (4 perfusions espacées de 2 mois chacune, associé à octréotide 30 mg tous les 28 jours) prolongeait la SSP en comparaison à l’octréotide double dose (60 mg tous les 28 jours) (SSP médiane 28,4 mois vs. 8,5 mois ; HR 0,21 ; p< 0,0001), chez des patients ayant une TNEG métastatique et progressive sous analogues de la SST simple dose (26). La RIV au 177Lu-DOTATATE est donc un traitement de référence après échec des analogues de la SST, en cas d’expression forte et homogène des récepteurs de la somatostatine, notamment en cas de métastases extra-hépatiques significatives.
La RIV peut également être envisagée en première ligne thérapeutique, en cas de volume tumoral important (en particulier si extra-hépatique) et/ ou de symptômes non contrôlés et/ou de TNEG de grade 2 ou 3, en association avec les analogues de la SST. En effet, dans cette indication l’étude NETTER-2 a montré que le 177Lu-DOTATATE (+ octréotide 30 mg/28 jours) prolongeait la SSP en comparaison à l’octréotide 60 mg/28 jours (SSP médiane 22,8 mois vs. 8,5 mois ; p< 0,0001) (27).
Thérapies ciblées orales
L’évérolimus est un inhibiteur de mTOR, protéine-clé de la régulation de la prolifération et le métabolisme des cellules tumorales. Dans l’étude de phase III RADIANT-4, l’évérolimus prolongeait la SSP en comparaison au placebo (médiane 11,0 mois vs. 3,9 mois, HR 0,48, p< 0,00001), chez des patients ayant une TNE non pancréatique (TNEG 31 %, pulmonaire 31 %, côlon-rectum 16 %) non fonctionnelle, progressive et ayant reçu au maximum une ligne de traitement systémique antérieur (28). Ainsi, l’évérolimus est indiqué dans les TNEG métastatiques progressives, notamment en cas de maladie extra-hépatique et/ou de négativité de l’expression des récepteurs à la somatostatine. Son profil de tolérance est néanmoins particulier, et son utilisation doit faire appel à certaines précautions spécifiques (29).
Le cabozantinib est un inhibiteur oral de protéines kinases impliquées dans l’angiogenèse (VEGF) et la prolifération (MET, AXL, RET) tumorale. Dans l’étude de phase III CABINET, le cabozantinib permettait de prolonger la SSP en comparaison au placebo (SSP médiane 8,4 vs. 3,9 mois, HR 0,38, p< 0,001), chez 203 patients ayant une TNE extra-pancréatique (TNEG 57 %) métastatique, surtout G2-G3 (70 %) et prétraitée (2 lignes thérapeutiques antérieures en moyenne), avec des effets secondaires fréquents et parfois significatifs mais le plus souvent gérables (30).
Chimiothérapie systémique
Les TNEG étant des tumeurs très peu chimio-sensibles, la chimiothérapie ne peut pas être recommandée en première intention, mais peut être utilisée comme traitement ultérieur après échec des autres options, notamment en cas de maladie rapidement progressive et/ou symptomatique. Alors que la chimiothérapie alkylante (à base de streptozotocine, témozolomide ou dacarbazine) est une référence dans les TNE pancréatiques, son efficacité est faible dans les TNEG probablement du fait d’une forte expression des enzymes de réparation des lésions de l’ADN induites par les alkylants (O6-méthylguanine-DNA méthyltransferase ou MGMT). Les meilleurs taux de réponse obtenus avec la chimiothérapie dans les TNEG concernent les associations de la capécitabine ou du FOLFOX au bévacizumab, anti-angiogénique (31,32).
Surveillance post-thérapeutique des TNEG
L’objectif de la surveillance des TNEG opérées à visée curative est de détecter précocement la récidive qui est généralement métastatique (ganglionnaire, hépatique ou péritonéale), afin de pouvoir proposer précocement un traitement efficace. Les TNEG étant habituellement de croissance lente, le patient opéré doit être informé de la possibilité de récidive tardive, et de la nécessité d’une surveillance prolongée pendant au moins 10 ans. Les modalités de suivi dépendent des facteurs pronostiques, comme le stade tumoral, le grade, l’index Ki-67 et le statut ganglionnaire. Il est recommandé de refaire un examen clinique, un dosage des marqueurs biochimiques et une imagerie morphologique (scanner, ou préférentiellement IRM car plus sensible pour la détection des petites métastases hépatiques) dans les 3 à 6 mois suivant la chirurgie, associée à l’imagerie nucléaire initialement positive (idéalement TEP/TDM à la 18FDOPA, sinon au 68Ga-DOTATOC), puis une imagerie morphologique tous les 6 à 12 mois pendant 5 ans, puis tous les 1 à 2 ans (2).
La surveillance des patients avec maladie en place traitée par traitement antitumoral dépend également des caractéristiques pronostiques tumorales et de l’efficacité du traitement. En fonction de ces critères, la surveillance est adaptée au cas par cas, en à un rythme généralement compris entre tous les 3 mois et tous les 6 mois. La réalisation systématique d’examens de médecine nucléaire n’a jamais démontré son utilité et ne permet pas de se passer d’examens réguliers d’imagerie radiologique (scanner, IRM) ; les TEP/TDM doivent donc être réservées à la confirmation d’une suspicion de progression ou de récidive équivoque en imagerie.
Références
Rindi G, Mete O, Uccella S, et al. Overview of the 2022 WHO Classification of Neuroendocrine Neoplasms. Endocr Pathol 2022;33:115–54. https://doi.org/10.1007/s12022-022-09708-2.
de Mestier L, Lepage C, Baudin E, et al. Digestive Neuroendocrine Neoplasms (NEN): French Intergroup clinical practice guidelines for diagnosis, treatment and follow-up (SNFGE, GTE, RENATEN, TENPATH, FFCD, GERCOR, UNICANCER, SFCD, SFED, SFRO, SFR). Dig Liver Dis 2020;52:473–92. https://doi.org/10.1016/j.dld.2020.02.011.
Grozinsky-Glasberg S, Davar J, Hofland J, et al. European Neuroendocrine Tumor Society (ENETS) 2022 Guidance Paper for Carcinoid Syndrome and Carcinoid Heart Disease. J Neuroendocrinology 2022:e13146. https://doi.org/doi.org/10.1111/jne.13146.
Lamarca A, Bartsch DK, Caplin M, et al. European Neuroendocrine Tumor Society (ENETS) 2024 guidance paper for the management of well-differentiated small intestine neuroendocrine tumours. J Neuroendocrinology 2024;36:e13423. https://doi.org/10.1111/jne.13423.
Dasari A, Shen C, Halperin D, et al. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncology 2017;3:1335–42. https://doi.org/10.1001/jamaoncol.2017.0589.
White BE, Rous B, Chandrakumaran K, et al. Incidence and survival of neuroendocrine neoplasia in England 1995–2018: A retrospective, population-based study. Lancet Reg Health Eur 2022;23:100510. https://doi.org/10.1016/j.lanepe.2022.100510.
Rinzivillo M, Capurso G, Campana D, et al. Risk and Protective Factors for Small Intestine Neuroendocrine Tumors: A Prospective Case-Control Study. Neuroendocrinology 2016;103:531–7. https://doi.org/10.1159/000440884.
Nuñez-Valdovinos B, Carmona-Bayonas A, Jimenez-Fonseca P, et al. Neuroendocrine Tumor Heterogeneity Adds Uncertainty to the World Health Organization 2010 Classification: Real-World Data from the Spanish Tumor Registry (R-GETNE). The Oncol 2018;23:422–32. https://doi. org/10.1634/theoncologist.2017-0364.
Strosberg J. Neuroendocrine tumours of the small intestine. Best Pract Res Clin Gastroenterol 2012;26:755–73. https://doi.org/10.1016/j. bpg.2012.12.002.
Basuroy R, Bouvier C, Ramage JK, Sissons M, Srirajaskanthan R. Delays and routes to diagnosis of neuroendocrine tumours. BMC Cancer 2018;18:1122. https://doi.org/10.1186/s12885-018-5057-3.
Lawrence B, Gustafsson BI, Kidd M, Pavel M, Svejda B, Modlin IM. The Clinical Relevance of Chromogranin A as a Biomarker for Gastroenteropancreatic Neuroendocrine Tumors. Endocrinology and Metabolism Clinics of North America 2011;40:111–34. https://doi. org/10.1016/j.ecl.2010.12.001.
de Mestier L, Savagner F, Brixi H, et al. Plasmatic and Urinary 5-Hydroxyindolacetic Acid Measurements in Patients With Midgut Neuroendocrine Tumors: A GTE Study. The Journal of Clinical Endocrinology & Metabolism 2021;106:e1673–82. https://doi.org/10.1210/clinem/ dgaa924.
Ronot M, Cuccioli F, Dioguardi Burgio M, et al. Neuroendocrine liver metastases: Vascular patterns on triple-phase MDCT are indicative of primary tumour location. European Journal of Radiology 2017;89:156–62. https://doi.org/10.1016/j.ejrad.2017.02.007.
d’Assignies G, Fina P, Bruno O, et al. High sensitivity of diffusion-weighted MRI for the detection of liver metastases from neuroendocrine tumors compared with T2-weighted and dynamic gadolinium-enhanced MRI, using histological findings as a standard of reference. Radiology 2013;268:390–9. https://doi.org/10.1148/radiol.13121628.
Singh S, Poon R, Wong R, Metser U. 68Ga PET Imaging in Patients With Neuroendocrine Tumors: A Systematic Review and Meta-analysis. Clin Nucl Med 2018;43:802–10. https://doi.org/10.1097/RLU.0000000000002276.
Ouvrard E, Chevalier E, Addeo P, et al. Intraindividual comparison of 18 F-FDOPA and 68 Ga-DOTATOC PET/CT detection rate for metastatic assessment in patients with ileal neuroendocrine tumours. Clin Endocrinol (Oxf) 2021;94:66–73. https://doi.org/10.1111/cen.14312.
Modlin IM, Gustafsson BI, Pavel M, Svejda B, Lawrence B, Kidd M. A nomogram to assess small-intestinal neuroendocrine tumor (‘carcinoid’) survival. Neuroendocrinology 2010;92:143–57. https://doi.org/10.1159/000319784.
de Mestier L, Hentic O, Brixi H, Ruszniewski P, Cadiot G. Antiproliferative effect of somatostatin analogs in neuroendocrine tumors. Hépato-Gastro & Oncologie Digestive 2014;21:835–44. https://doi.org/10.1684/hpg.2014.1090.
Lardière-Deguelte S, de Mestier L, Appéré F, et al. Toward Preoperative Classification of Lymph-Node Metastases in Patients with Small Intestine Neuroendocrine Tumours in the Era of Intestinal-Sparing Surgery. Neuroendocrinology 2015. https://doi.org/10.1159/000441423.
Frilling A, Modlin IM, Kidd M, et al. Recommendations for management of patients with neuroendocrine liver metastases. Lancet Oncol 2014;15:e8-21. https://doi.org/10.1016/S1470-2045(13)70362-0.
de Mestier L, Lardière-Deguelte S, Brixi H, et al. Updating the surgical management of peritoneal carcinomatosis in patients with neuroendocrine tumors. Neuroendocrinology 2015;101:105–11. https://doi.org/10.1159/000371817.
Rinke A, Müller H-H, Schade-Brittinger C, et al. Placebo-Controlled, Double-Blind, Prospective, Randomized Study on the Effect of Octreotide LAR in the Control of Tumor Growth in Patients With Metastatic Neuroendocrine Midgut Tumors: A Report From the PROMID Study Group. JCO 2009;27:4656–63. https://doi.org/10.1200/JCO.2009.22.8510.
Caplin ME, Pavel M, Cwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med 2014;371:224–33. https://doi.org/10.1056/NEJMoa1316158.
Pavel M, Ćwikła JB, Lombard-Bohas C, et al. Efficacy and safety of high-dose lanreotide autogel in patients with progressive pancreatic or midgut neuroendocrine tumours: CLARINET FORTE phase 2 study results. European Journal of Cancer 2021;157:403–14. https://doi.org/10.1016/j. ejca.2021.06.056.
de Mestier L, Zappa M, Hentic O, Vilgrain V, Ruszniewski P. Liver transarterial embolizations in metastatic neuroendocrine tumors. Rev Endocr Metab Disord 2017;18:459–71. https://doi.org/10.1007/s11154-017-9431-2.
Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N Engl J Med 2017;376:125–35. https://doi.org/10.1056/NEJMoa1607427.
Singh S, Halperin D, Myrehaug S, et al. [177Lu]Lu-DOTA-TATE plus long-acting octreotide versus high-dose long-acting octreotide for the treatment of newly diagnosed, advanced grade 2–3, well-differentiated, gastroenteropancreatic neuroendocrine tumours (NETTER-2): an open-label, randomised, phase 3 study. The Lancet 2024;403:2807–17. https://doi.org/10.1016/S0140-6736(24)00701-3.
Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet 2016;387:968–77. https://doi.org/10.1016/S0140-6736(15)00817-X.
Hentic O, de Mestier L. Évérolimus et sunitinib dans le traitement des tumeurs neuroendocrines digestives : utilisation en pratique. Hépato Gastro 2017;24:22–38. https://doi.org/10.1684/hpg.2017.1545.
Chan JA, Geyer S, Zemla T, et al. Phase 3 Trial of Cabozantinib to Treat Advanced Neuroendocrine Tumors. N Engl J Med 2025;392:653–65. https://doi.org/10.1056/NEJMoa2403991.
Lacombe C, Perrier M, Hentic O, et al. FOLFOX-bevacizumab chemotherapy in patients with metastatic neuroendocrine tumors. J Neuroendocrinol 2023;35:e13227. https://doi.org/10.1111/jne.13227.
Mitry E, Walter T, Baudin E, et al. Bevacizumab plus capecitabine in patients with progressive advanced well-differentiated neuroendocrine tumors of the gastro-intestinal (GI-NETs) tract (BETTER trial) – A phase II non-randomised trial. Eur J Cancer 2014;50:3107–15. https://doi.org/10.1016/j.ejca.2014.10.001.
Toute reproduction ou réécriture, totale ou partielle, sans l’accord préalable écrit de la FMC HGE est interdite.
FMC HGE : Organisme certifié Qualiopi pour la catégorie ACTIONS DE FORMATION.
Nous vous invitons à tester vos connaissances sur l’ensemble des QCU tirés des exposés des différents POST’U. Les textes, diaporamas ainsi que les réponses aux QCM seront mis en ligne à l’issue des prochaines journées JFHOD.