Introduction
La cholestase est définie par la diminution de la formation de la bile (ou cholérèse), une fonction hépatique non directement mesurable en pratique. Cependant, des signes cliniques ou biologiques faciles à recueillir sont de sensibilité ou spécificité élevées pour le diagnostic de cholestase. De ce fait, la notion de cholestase est probablement l’une des entités sémiologiques les plus précieuses dont dispose l’hépa- tologue. Cet exposé et le texte ci-dessous actualiseront l’exposé donné au séminaire de la SNFGE de novembre 2013 ainsi que l’article correspondant intitulé « Cholestase » publié dans HépatoGastro et Oncologie Digestive en septembre 2013 (1).
La cholérèse
La bile
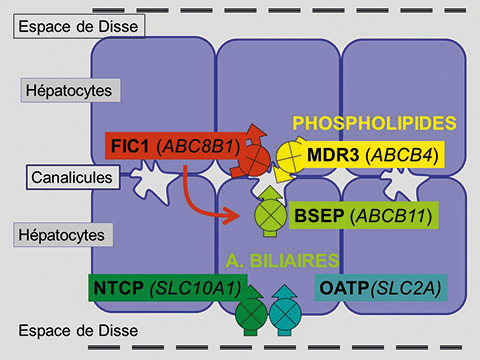
La bile est un liquide aqueux (97 % d’eau) assurant l’écoulement jusqu’au duodénum de multiples substances produites par le foie ou les voies biliaires elles-mêmes. La production de bile est assurée principalement par la sécrétion d’acides biliaires au pôle biliaire des hépatocytes (2-4). Ce pôle biliaire participe à la formation du canalicule (figure 1). Cette sécrétion, qui nécessite de l’énergie, est assurée par un transporteur nommé ABCB11 ou Bile Salt Export Pump (BSEP). Le pouvoir osmotique des acides biliaires crée un appel d’eau interstitielle, qui traverse les jonctions serrées intercellulaires des hépatocytes que les acides biliaires ne peuvent franchir.

Figure 1. Transporteurs hépatocytaires indispensables à la production de la bile par captation des acides biliaires au pôle sinusoïdal des hépatocytes et sécrétion au pôle canaliculaire. Une petite portion des acides biliaires est synthétisée quotidiennement dans l’hépatocyte à partir du cholestérol. ATP8B1, FIC1. ABCB11, BSEP. ABCB4, MDR3. SLC10A1, NTCP. SLC2A, OATP.
Un courant biliaire principal est ainsi formé.
Il s’y ajoute dans le canalicule, le petit courant hydrique, dit « fraction du débit biliaire indépendante des acides biliaires », généré par la sécrétion d’autres substances hydrosolubles dans le canalicule.
Un peu plus loin, dans l’arbre biliaire tapissé de cellules biliaires ou cholangiocytes, une autre fraction hydrique est ajoutée au courant principal (4-6). Cette dernière fraction, qui compte pour 25 à 30 % du débit biliaire, naît de la sécrétion des ions chlore et bicarbonate au pôle biliaire des cholangiocytes, par le transporteur CFTR (ABCC7) pour le chlore, par l’échangeur d’anions chlore-bicarbonates et par des canaux spécifiques pour le passage transmembranaire de l’eau (aquaporines). Les mutations de CFTR, responsables de la mucoviscidose, peuvent effectivement s’accompagner d’une atteinte biliaire. La sécrétion de bicarbonates permet la mise en place d’un « parapluie » alcalin qui protège les acides biliaires d’une protonation (5). En effet, les acides biliaires protonés peuvent pénétrer dans les cholangiocytes et y provoquer des lésions.
Le débit normal quotidien de bile est de l’ordre de 700 ml/jour. Il est augmenté par la sécrétine qui agit sur la fraction cholangiocytaire du débit biliaire.
Acides biliaires
Les acides biliaires conjugués, synthétisés par l’hépatocyte à partir du cholestérol, sont l’objet d’une recirculation intensive dans le cycle entérohépatique (3, 6). Ce cycle fait intervenir 2 pompes mécaniques, la vésicule biliaire et l’intestin. Dans l’intestin, les acides biliaires déconjugués par les bactéries sont réabsorbés passivement. Ces 2 pompes mécaniques sont séparées par 3 pompes chimiques : i) ABCB11 (BSEP), déjà envisagée, pour la sécrétion canaliculaire ; ii) SLC10A2 (solute carrier transporter A2 ou apical sodium-dependent bile salt transporter ou ASBT) du pôle luminal des entérocytes de l’iléon terminal, couplé à OSTa/b au pôle portal, pour la réabsorption quasi complète des acides biliaires intestinaux ; et iii) SLC10A1 (ou sodium taurocholate cotransporter protein, NTCP) et SLC2A3/6/8 (ou organic anion-transporting polypeptide OATP -/2/8), pour la captation les acides biliaires au pôle sinusoïdal des hépatocytes.
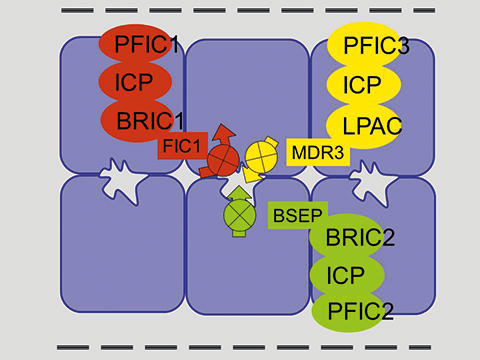
Les mutations qui inactivent ABCB11 (BSEP) sont responsables de la cholestase familiale progressive de type 2 (ou PFIC2 pour progressive familial intrahepatic cholestasis type 2) et de la cholestase récurrente bénigne (ou BRIC2 pour benign recurrent intrahepatic cholestasis type 2) (Figure 2) (7, 8).
Figure 2. Les anomalies génétiques responsables de cholestase par atteinte des transporteurs canaliculaires et leur expression clinique. PFIC1, 2, 3, cholestase familiale progressive de type 1, 2, 3. BRIC1, 2, cholestase récurrente bénigne de type 1, 2. ICP cholestase gravidique. LPAC, low phospholipid associated cholestasis and cholelithiasis.
Bilirubine
La bilirubine ne participe pas ou peu à la formation du courant hydrique de la bile, mais elle met ce courant à profit pour son élimination vers l’intestin (9). En pratique, la bilirubine produite par la dégradation de l’hème est principalement issue des globules rouges, en raison du fort taux de renouvellement de ces cellules comparé aux autres tissus également riches en hème (muscles pour la myoglobine, foie pour les cytochromes, par exemple). La bilirubine non conjuguée, liposoluble, est liée de façon équimolaire à l’albumine dans le sang. Elle franchit la membrane sinusoïdale des hépatocytes par simple diffusion. Dans le cytoplasme des hépatocytes, elle est liée à la glutathion S-transférase (ou ligandine). La bilirubine est conjuguée à l’acide glucuronique par l’UDP glucuronyl bilirubine transferase (ou UGT1A1) du réticulum endoplasmique. Une fois conjuguée, la bilirubine, hydrosoluble, peut être sécrétée dans le canalicule par un transporteur polyvalent, ABCC2, prenant en charge les anions organiques conjugués (comme la bilirubine conjuguée, le glutathion, ou certains acides biliaires conjugués, mais aussi des xénobiotiques comme la BSP ou certains anticancéreux, d’où son ancien nom de multidrug resistance associated protein 2 (ou MRP2). Les mutations inactivant le gène de ABCC2 expliquent le très rare syndrome de Dubin-Johnson qui se manifeste par une augmentation isolée de la bilirubinémie conjuguée.
La bilirubine conjuguée de l’hépatocyte peut aussi être sécrétée dans les sinusoïdes par un autre transporteur, ABCC3. La bilirubine conjuguée du sang sinusoïdal peut être captée à nouveau par l’hépatocyte, au moyen de transporteurs polyvalents pour de nombreux anions organiques (organic anion transporting polypeptide 1 B1 ou B3, OATP1 B1/B3). Le syndrome de Rotor est une anomalie très rare due à des mutations inactivant OATP1 B1/B3. Il est caractérisé par une augmentation dans le sérum à la fois de la bilirubine conjuguée et de la bilirubine non conjuguée (10).
Les mutations affectant directement le métabolisme de la bilirubine, mais non le métabolisme des acides biliaires ne déterminent pas de cholestase. À l’exception notable de la maladie de Crigler-Najjar, elles sont bénignes (10, 11).
Phospholipides biliaires
Il s’agit principalement de la phosphatidyl-choline (ou lécithine). Constituant normal de la membrane cellulaire, la phosphatidyl-choline en déplacée du feuillet interne de la membrane canaliculaire vers le feuillet externe bile par le transporteur ABCB4 (encore nommé multidrug resistance protein 3 ou MDR3), ce qui autorise sa sécrétion dans la bile (figure 1). Tous deux amphiphiles, les acides biliaires et la phosphatidyl-choline entrent dans la composition de micelles mixtes où le cholestérol est solubilisé.
Lorsque le rapport de concentration entre phosphatidyl-choline, acides biliaires et cholestérol n’est pas approprié, le cholestérol insoluble dans l’eau précipite sous forme de cristaux dans les petites voies biliaires. C’est ce que décrit le fameux triangle de Small (12). L’augmentation de volume des cristaux mène à la formation des calculs biliaires macroscopiques. Les mutations inactivant le gène ABCB4 sont à l’origine du syndrome de la bile pauvre en phosphatidyl-choline (low phospholipid associated cholestasis and cholelithiasis, ou LPAC) (13). Ce syndrome peut comporter une atteinte des petites voies biliaires attribuée à l’effet détergent des acides biliaires normalement contrôlé par leur incorporation dans les micelles mixtes.
La membrane canaliculaire est également le siège d’un transporteur de phospholipides nommé ATP8B1 (ou familial intrahepatic cholestasis type 1 protein, FIC1), ce transporteur de type P-ATPase est inactivé par les mutations responsables de la Maladie de Byler (ou cholestase familiale progressive de type 1) et de la cholestase récurrente bénigne de type 1 (ou BRIC1 pour benign recurrent intrahepatic cholestasis type 1) (figure 2) (8, 14, 15). Il s’agit d’un transporteur des aminophospholipides (phosphatidyl-sérine et phosphatidyl-éthanolamine) du feuillet externe au feuillet interne de la membrane cellulaire. Son implication exacte dans les transports biliaires et dans le développement de la cholestase familiale ne sont pas parfaitement connus. Il semble indispensable au maintien de la membrane canaliculaire dans un état de résistance à l’action détergente des acides biliaires.
Cholestérol
Le cholestérol biliaire, non estérifié, est synthétisé par le foie. Il compte pour 4 fois plus que les apports alimentaires normaux dans le débit intestinal quotidien du cholestérol. Le passage du cholestérol, hydrophobe, dans la bile est sous la dépendance de transporteurs canaliculaires (ABCG5/8) (4, 8). Il est aussitôt solubilisé dans les micelles (par les acides biliaires), les micelles mixtes et les vésicules unilamellaires, (avec les phospholipides). La moitié du cholestérol biliaire est réabsorbé dans l’intestin.
Bicarbonates
Les bicarbonates sont les principaux cations de la bile. Ils lui confèrent son caractère alcalin et expliquent le rôle de « parapluie » évoqué plus haut (5).
Protéines
De nombreuses protéines de divers types enzymatiques, ou non, sont présentes dans la bile. Parmi celles-ci, les mucines sont impliquées dans la formation des calculs biliaires. Les IgA polymériques circulantes sont captées par endocytose au pôle sinusoïdal par une liaison avec le composant sécrétoire membranaire ; transportées à travers l’hépatocyte jusqu’au pôle canaliculaire ; et déversée dans la bile avec une partie du composant sécrétoire sous forme d’IgA sécrétoire.
Diagnostic d’une cholestase chronique
Le syndrome de cholestase inclut de nombreux signes cliniques et biologiques différents, rassemblés dans le tableau 1. Les signes cliniques les plus importants sont l’ictère à bilirubine conjuguée envisagé plus haut et le prurit. Le prurit cholestatique n’est initialement associé à aucune lésion cutanée, ce qui n’est pas spécifique : de nombreuses autres affections non cutanées peuvent être en cause. D’autre part, les lésions de la peau secondaire au prurit chronique sont fréquentes et variées : lichénification, nodules de prurigo, plaques de dermatite et excoriations dues au grattage. Le prurit est lié à une atteinte de la transmission nerveuse encore mal caractérisée (16). L’association du prurit à une augmentation de l’autaxine dans le sérum a maintenant été bien démontrée, mais n’est pas expliquée (17). L’autaxine catalyse la formation d’acide lysyl-phosphatidique qui pourrait être le médiateur de la stimulation nerveuse conduisant au prurit.
Les signes biologiques les plus importants sont l’augmentation de l’activité phosphatase alcaline du sérum et de la bilirubine conjuguée.
| Signes cliniques | Signes biologiques (sérum ou plasma) |
|---|
| Ictère à bilirubine conjuguée Prurit Amaigrissement, stéatorrhée Signes de carence en vitamines A,D,E,K (cholestase prolongée) | Augmentation des acides biliaires Augmentation de l’activité phosphatase alcaline Augmentation de l’activité gamma-glutamyl transpeptidase Augmentation de l’activité 5’ nucléotidase Augmentation de l’activité des transaminases ALAT et ASAT (cholestase aiguë) Taux de Quick diminué et facteur V normal (cholestase chronique) |
Tableau 1. Principaux éléments cliniques et biologiques du syndrome de cholestase
Phosphatases alcalines
L’augmentation de l’activité phosphatase alcaline du sérum est l’élément clé du syndrome de cholestase. Les phosphatases alcalines sont des enzymes localisées à la membrane plasmique. Le site actif est orienté du côté de la lumière. Elles sont ancrées dans la membrane par un groupement GPI. Leur activité consiste à cliver un groupement phosphate et leurs substrats comprennent un grand nombre de substance (protéines, nucléotides). Il en existe 3 isoformes, chacune codée par un gène différent : celle du placenta, celle de l’intestin et une forme non-spécifique de tissu, trouvée en grande quantité dans les tissus ayant des fonctions d’échange ou de transport soutenues comme le rein, l’intestin et le foie. Dans le foie normal, elles sont localisées particulièrement au voisinage du canalicule biliaire et dans la membrane plasmique des cholangiocytes.
Cette activité enzymatique peut augmenter dans le sérum dans 3 principales circonstances : la cholestase, la fin de la grossesse (elle est d’origine placentaire) et l’augmentation de la construction osseuse. Cette dernière peut paradoxalement s’associer aux lésions ostéolytiques. Au cours de la cholestase, l’expression des phosphatases alcalines augmente au niveau de la membrane du pôle sinusoïdal des hépatocytes et dans le cytoplasme des cholangiocytes. L’augmentation dans le sérum pourrait résulter à la fois de cette induction et de cette redistribution des phosphatases alcalines à la membrane plasmique du pôle sinusoïdal et aussi de sa désinsertion de la membrane sous l’effet détergent des acides biliaires en excès. Au cours de la cholestase, l’augmentation des phosphatases alcalines n’est pas brutale et le retour à une activité normale est lent après la levée de la cause (18). Ce fait et d’autres arguments indiquent qu’il ne s’agit pas d’un simple passage par régurgitation de constituants biliaires dans le sérum.
Gamma-glutamyl transferase (GGT)
Cette enzyme est également localisée dans la membrane des cellules d’un grand nombre de tissus incluant le foie, le pancréas, le rein et les voies biliaires. Sa fonction est de transférer un résidu gamma-glutamyl à un autre peptide ou acide aminé. Le substrat le plus abondant est le glutathion.
Toutes les atteintes hépatiques et biliaires peuvent augmenter l’activité sérique de cette enzyme et ce de façon parfois très marquée. L’augmentation de la GGT est un test assez sensible de consommation excessive d’alcool ou de syndrome métabolique. Comme ces dernières circonstances sont fréquentes dans la population générale, l’augmentation de la GGT manque totalement de spécificité pour le diagnostic de cholestase. Au cours de la cholestase, l’augmentation de la GGT est fortement associée à la prolifération ductulaire (ou cholangiolaire) que l’on observe en cas d’obstruction ou de destruction des voies biliaires, mais qui manque en cas de cholestase par atteinte canaliculaire (19). L’augmentation de l’activité sérique résulte probablement aussi, comme pour les phosphatases alcalines, d’une libération de la membrane cellulaire par l’action détergente des acides biliaires (19).
Le principal intérêt de la détermination de l’activité de la GGT sérique est d’attribuer une augmentation des phosphatases alcalines à une maladie du foie ou des voies biliaires et non pas à une atteinte osseuse. On doit prendre garde à ne pas conclure par excès à une cholestase en cas d’augmentation isolée de la GGT ; une telle hypothèse ne peut être retenue en l’absence d’une cause claire d’atteinte biliaire. Le second intérêt de la détermination de la GGT en cas de cholestase naît d’un résultat normal ou très peu élevé. En effet, cela suggère l’absence de prolifération ductulaire. Ce type de cholestase sans prolifération ductulaire est rencontré en cas de cholestase familiale progressive ou de cholestase récurrente bénigne de type 1 (dues à une atteinte d’ATP8B1) ou de type 2 (dues à une atteinte de ABCB11) (15).
Bilirubinémie
La bilirubine non conjuguée, liée à l’albumine, ne peut être filtrée dans le globule rénal. La bilirubine conjuguée est éliminée par filtration glomérulaire et réabsorption et sécrétion tubulaire. Sa clairance plasmatique est d’environ la moitié de celle de la créatinine. Chez le sujet normal, la concentration sérique de bilirubine totale est inférieure à 17µmol/L ; les ¾ de la bilirubine sérique sont non conjugués et ¼ est conjugué à l’acide glucuronique. L’augmentation de la bilirubine dans le sérum est visible par l’œil au niveau des parties claires des téguments (peau blanche et sclérotique) sous forme d’un ictère lorsque la concentration de la bilirubine dépasse 40 µmol/L. En cas d’ictère, des urines claires indiquent qu’il s’agit principalement d’une augmentation de la bilirubine non conjuguée. Des urines très foncées indiquent une hyperbilirubinurie conjuguée, due à une hyperbilirubinémie en l’absence de concentration marquée des urines.
Une hyperbilirubinémie non conjuguée ne peut s’expliquer par une cholestase (9). À l’inverse, en théorie, une hyperbilirubinémie conjuguée ne signifie pas nécessairement une cholestase : les syndromes de Rotor et de Dubin-Johnson peuvent l’expliquer par un simple trouble du transport de la bilirubine. En pratique, l’extrême rareté de ces 2 syndromes rend l’hyperbilirubinémie conjuguée synonyme d’une cholestase (9). Une cholestase peut être présente sans ictère, puisque le métabolisme de la bilirubine est (relativement) indépendant de celui des acides biliaires qui, lui, est directement en cause dans la cholestase. En fait, la présence ou l’absence d’ictère pour un degré similaire de cholestase n’est pas toujours facile à expliquer. En cas d’ictère sans obstruction biliaire complète, cette discordance s’explique probablement par l’association de plusieurs facteurs à la cholestase ; par exemple : i) une hyperhémolyse associée constitutionnelle ou acquise qui augmente la charge en bilirubine ; ii) un défaut associé constitutionnel (polymorphisme génétique) ou acquis (inflammation) des transporteurs propres de la bilirubine ; ou iii) une insuffisance rénale qui diminue la clairance plasmatique de la bilirubine conjuguée.
Les médicaments, utilisant les mêmes voies métaboliques que la bilirubine, peuvent provoquer un ictère à bilirubine non conjuguée, par simple compétition. C’est en particulier le cas de certains inhibiteurs de protéase du virus de l’immunodéficience humaine (9).
Excepté en cas d’atteinte de la glucuro-conjugaison (syndromes de Gilbert ou de Crigler-Najjar), une hyperbilirubinémie non conjuguée est toujours associée à une hyperbilirubinémie conjuguée (9). La charge en bilirubine non conjuguée pourrait en effet induire i) une augmentation l’efflux de bilirubine conjuguée par ABCC3 de la membrane sinusoïdale, et/ou ii) une saturation de l’excrétion canaliculaire de la bilirubine conjuguée alors que la limite de saturation de la glucuro-conjugaison n’est pas atteinte. On estime que l’anomalie porte principalement sur le métabolisme de la bilirubine conjuguée lorsque la fraction de cette dernière dans le sérum est supérieure à 75 % du total de la bilirubinémie (ce qui correspond approximativement à la proportion normale).
Autres éléments du syndrome de cholestase
L’apport d’autres déterminations que celle des phosphatases alcalines, de la GGT et de la bilirubine pour la prise en charge diagnostique, pronostique et thérapeutique d’un syndrome de cholestase est pratiquement nulle. La valeur pronostique de la détermination des concentrations sériques d’acide biliaire au cours de la cholestase gravidique est de peu d’utilité en pratique excepté dans les rares cas où elle est supérieure à 100 µmol/L.
Syndrome de cholestase chronique
Il n’y a pas de définition aussi claire de la chronicité pour la cholestase que pour l’hépatite. C’est pourquoi la notion de cholestase prolongée est plus souvent utilisée que celle de cholestase chronique. Plus que la durée, c’est le caractère symptomatique ou non et le caractère abrupt ou non du début des manifestations de cholestase qui a un intérêt clinique. Les affections sources de cholestase de durée spontanément limitée (souvent dites aiguës) sont les hépatites médicamenteuses ou virales cholestatiques ; les obstructions transitoires ou intermittentes des voies biliaires par des calculs ; les épisodes de cholestase récurrente bénigne ; la cholestase satellite d’une infection sévère. Les épisodes de cholestase de début brutal sont souvent associés à une augmentation marquée, mais transitoire des transaminases, alors que cette augmentation est fréquente, mais peu marquée en cas de cholestase chronique d’ins- tallation progressive, même si elle est finalement d’un degré très prononcé.
Diagnostic du syndrome de cholestase chronique
Un diagnostic de cholestase peut être porté i) lorsqu’un prurit ou un ictère à bilirubine conjuguée s’associent à une augmentation des phosphatases alcalines ; ou ii) lorsqu’une une augmentation des phosphatases alcalines s’associe à une augmentation de la GGT. Dans ce dernier cas, le diagnostic est conforté par l’absence de consommation importante d’alcool et de syndrome métabolique, ou par la présence d’une cause possible de cholestase.
Une cholestase bien démontrée avec des phosphatases alcalines normales doit faire évoquer une forme mineure d’aphosphatasie. Comme indiqué ci-dessus, une cholestase sans augmentation de la GGT est évocatrice d’une atteinte génétique d’ATP8B1 ou ABCB11. En l’absence d’histoire familiale, de prurit, ou d’ictère, le diagnostic en est très difficile.
La couleur des urines, la détermination des phosphatases alcalines et la détermination des fractions conjuguées et non conjuguées de la bilirubine rend facile le diagnostic différentiel de l’ictère cholestatique.
Étiologie du syndrome de cholestase
Lorsque les phosphatases alcalines sont supérieures à 3 fois la valeur supérieure de la norme, la discussion étiologique doit débuter par celle d’une cholestase, quelle que soit la valeur des transaminases. En revanche, en dessous de ce seuil et en l’absence de douleur biliaire, il est probablement plus approprié de débuter par une discussion de l’augmentation des transaminases lorsqu’elles sont supérieures à 5 fois leur limite normale supérieure.
Une classification des causes de cholestase opérationnelle en clinique est bien connue de tous. Elle est résumée dans le tableau 2. La principale dichotomie est constituée par l’atteinte ou le respect des voies biliaires.
Tableau 2. Classification des causes de cholestase| Avec atteintes des voies biliaires | Sans atteinte des voies biliaires |
|---|
| Grosses voies biliaires | Petites voies biliaires | Atteinte des transporteurs canaliculaires |
|---|
| Obstacle endoluminal Tumeur - Tumeur bénigne
- Cancer des voies biliaires, du pancréas, cystadénocarcinome, métastase endobiliaire
Sténose bénigne - Cholangite sclérosante primitive
- Cholangite à IgG4/pancréatite autoimmune
- Sténose post opératoire
| - Cirrhose biliaire primitive
- Cholangite immunoallergique
- Ductopénie idiopathique
- Syndrome d’Alagille
- Syndrome LPAC*, PFIC3* (ABCB4)
- Mucoviscidose
| Atteintes génétiques- PFIC* (ATP8B1, ABCB11, ABCB4)
- BRIC* (FIC1, BSEP)
- ICP* (FIC1, BSEP, MDR3)
Atteintes acquises - Inflammation/infection
- systémique
- hépatite aiguë (virale, autoimmune, immunoallergique)
- cirrhose
- Xenobiotiques
|
| *PFIC (pour Progressive familial intrahepatic cholestasis), cholestase familiale progressive. BRIC (pour benign recurrent intrahepatic cholestasis), cholestase récurrente bénigne. ICP (pour intrahepatic cholestasis of pregnancy), cholestase gravidique. LPAC (pour low phospholipid associated cholestasis and cholelithiasis), syndrome de la bile pauvre en phospholipides. |
Atteinte des voies biliaires
La cholestase peut résulter d’une atteinte des gros canaux (c’est-à-dire visualisable par imagerie) ; ou d’une atteinte des canaux biliaires de petit ou moyen calibre (observables seulement par examen microscopique d’une biopsie hépatique). Une atteinte limitée de la voie biliaire principale peut déterminer un ictère. En revanche, une atteinte des canaux de plus petit calibre doit être diffuse pour entraîner un ictère. L’obstruction d’un seul canal hépatique droit ou gauche ne suffit pas pour déterminer un ictère, alors que leur obstruction simultanée le peut.
Une douleur biliaire est générée par la mise en tension brutale des voies biliaires. De ce fait, lorsqu’un ictère survient à la suite d’une douleur biliaire, il est probable que l’obstacle s’est constitué brutalement. Ce mécanisme répond généralement à la migration d’un calcul dans la voie biliaire principale.
Une cholestase prolongée, quel qu’en soit le mécanisme, induit rapidement un amaigrissement marqué, en raison de la malabsorption qu’elle entraîne. De ce fait, un amaigrissement apparaissant après la constitution d’un ictère cholestatique n’a aucune spécificité. En revanche, un amaigrissement précédant la constitution d’un ictère cholestatique, doit faire envisager en premier lieu une autre cause d’amaigrissement, par exemple un cancer ou une affection inflammatoire chronique.
Quel que soit le siège de l’obstacle, une dilatation harmonieuse des voies biliaires est habituellement observée dans tout le territoire en amont de l’obstacle. Cependant, la dilatation peut manquer lorsque l’examen est fait peu après une obstruction récente (comme la migration d’un calcul dans la voie biliaire principale) ; ou lorsque les voies biliaires en amont ne peuvent se dilater, parce que leur paroi est scléreuse ; ou que le parenchyme hépatique est anormalement rigide (par exemple du fait d’une cirrhose).
Une dilatation de la vésicule biliaire indique à la fois que l’obstacle est cholédocien (en aval de la convergence du canal cystique et du canal hépatique commun) et que la vésicule est saine (non lithiasique).
Le cancer du pancréas, le cancer primitif de la voie biliaire principale et l’angiocholite par calcul de la voie biliaire principale sont les causes majeures d’ictère cholestatique. Des causes moins fréquentes sont la sténose post-opératoire des voies biliaires et la compression de la voie biliaire principale par une pancréatite chronique calcifiante ou par une adénopathie (tumorale ou inflammatoire). De nombreuses autres causes d’obstruction de la voie biliaire principale sont possibles ; chacune est très rare ; elles ne rendent compte, à elles toutes, que d’une minorité des cas de cholestase.
La cholangite sclérosante et la cholangite à IgG4 sont maintenant principalement reconnues par l’IRM biliaire montrant de façon assez peu spécifique une succession de sténoses et de dilatation. Les atteintes extra-biliaires (colite inflammatoire pour la cholangite sclérosante primitive et pancréatite autoimmune ou autre atteinte à IgG4 pour la cholangite à IgG4) sont des éléments majeurs du diagnostic. Le diagnostic différentiel avec un cholangiocarcinome peut s’avérer extrêmement difficile (20).
La cirrhose biliaire primitive est caractérisée par une cholangite destructrice non suppurée, affectant les canaux biliaires microscopiques. Les anticorps anti-mitochondries de type M2 ou certains anticorps anti- noyau y sont quasi-constants. L’association de ces autoanticorps et d’un syndrome de cholestase permet de se passer de la biopsie hépatique diagnostique (21).
Les cholangite immuno-allergiques sont principalement médicamenteuses. Les médicaments habituellement en cause sont : l’association acide clavulanique-amoxycilline, les sulfamides, les macrolides et l’allopurinol. Une fièvre, des douleurs marquées de l’hypochondre droit et une hyperéosinophilie y sont fréquentes (22).
Deux affections génétiques déjà mentionnées peuvent déterminer une atteinte des canaux biliaires de petit ou moyen calibre et conduire à une cholestase : la mucoviscidose et le syndrome LPAC ou PFIC3 due à une mutation de ABCB4 (5).
Cholestase sans obstacle sur les canaux biliaires
Les atteintes génétiques des transporteurs biliaires envisagées plus haut sont extrêmement rares. Elles produisent soit une cholestase infantile conduisant à une cirrhose (PFIC ou cholestase intrahépatique familiale progressive) ; soit des épisodes récidivants de cholestase spontanément régressive, imprévisibles en nombre et en durée, dont les facteurs déclenchant ne sont pas connus (cholestase récurrente bénigne) ; soit à une cholestase gravidique (8, 14, 15, 23).
Les atteintes acquises des transporteurs biliaires sont liées à une inhibition du transport des acides biliaires et de la bilirubine par les cytokines proinflammatoires (IL2, IL1, IL6) (24). Ce mécanisme explique l’ictère observé au cours des hépatites aiguës (alcoolique, virales, auto-immune ou médicamenteuses), même en l’absence de cholestase et d’insuffisance hépatique marquées. Ce mécanisme explique encore la cholestase ou l’ictère souvent observé au cours des infections bactériennes sévères (pyélonéphrite aiguë, pneumonie bactérienne, typhoïde, leptospirose, etc.). L’inflammation contribue à l’ictère dû à une angiocholite. Des interactions médicamenteuses sont également possibles (22).
Fréquemment, un ictère à bilirubine conjuguée ne relève pas d’un seul des mécanismes précédents mais d’une conjonction de différents facteurs. C’est le cas des malades dans un état grave, souvent infectés, atteints de cirrhose ou nécessitant des soins intensifs quelle qu’en soit la raison.
S’associent alors : une cholestase par inhibition de la captation sinusoïdale des acides biliaires, une diminution de la sécrétion canaliculaire de la bilirubine conjuguée due au syndrome inflammatoire ; une hyperhémolyse (due à des transfusions, des dispositifs intravasculaires, ou des anomalies érythrocytaires acquises) ; une insuffisance hépatique ; et une insuffisance rénale.
Diagnostic de la cause de la cholestase
La démarche est bien connue. Elle commence par l’échographie pour repérer des anomalies des voies biliaires. Celles-ci seront alors précisées par une cholangiopancréatographie par IRM. L’échoendoscopie permet parfois de préciser des points particuliers. Le cathétérisme rétrograde endoscopique des voies biliaires est un outil principalement thérapeutique. Lorsque l’échographie ne suggère pas d’anomalie des voies biliaires, le premier test doit être une recherche d’anticorps anti-mitochondries et d’anticorps antinucléaires spécifiques de la cirrhose biliaire primitive. Si l’atteinte est aiguë, une recherche des virus des hépatites est justifiée. La prise récente d’un médicament dont l’effet cholestatique est notoire doit le faire interrompre et suivre l’évolution des tests. Si ces différentes investigations sont négatives, il sera difficile de se passer d’une biopsie hépatique. Le diagnostic génétique ne permet que de préciser un diagnostic de cholestase familiale progressive, cholestase récurrente bénigne ou cholestase gravidique porté sur les autres arguments. L’interprétation des divergences de séquence génétique par rapport à la séquence de référence doit être très prudente.
Évolution des maladies chroniques du foie cholestatiques
Quelle qu’en soit la cause, les conséquences hépatiques d’une cholestase chronique sont une fibrose s’aggravant progressivement, débutant dans la région périportale pour finalement constituer une cirrhose particulière parce qu’elle peut laisser des régions centrolobulaires intactes au centre des lobules. Cette cirrhose (dite biliaire secondaire) est associée à une hypertension portale et à ses complications. Les complications attribuées à l’insuffisance hépatique (ascite, syndrome hépatorénal et encéphalopathie) sont plus tardives. L’hypoalbuminémie et la bilirubinémie sont des éléments pronostiques indépendants pour la plupart des cholestases chroniques sévères. La cirrhose biliaire secondaire est réversible lorsque la cause de la cholestase peut être complètement corrigée (comme par la levée d’un obstacle mécanique (25). À défaut d’être supprimée, lorsque la cause peut être contrôlée (par exemple par le traitement par acide ursodéoxycholique au cours de la CBP), la progression des lésions de fibrose est fortement ralentie, voire stoppée (26). Les complications hépatiques constituent la principale cause de décès au cours des cholestases chroniques réfractaires ou intraitables.
Manifestations extra-hépatiques des cholestases chroniques
Indépendamment des manifestations associées à la cause (qui peuvent être multiples en cas de maladie auto-immune par exemple), la cholestase entraîne des manifestations extra-hépatiques parfois sévères. Les troubles de la coagulation liés à la malabsorption de la vitamine K sont faciles à identifier par la mesure du facteur V dont le niveau est préservé alors que celui du taux de Quick est bas. Les troubles osseux liés à la cholestase chronique ont été appelés ostéodystrophie hépatique pour tenir compte qu’il s’agit tout autant d’une perte osseuse (ostéoporose) que d’un défaut de minéralisation. Elle se traduit par une perte de hauteur, des douleurs dorsales ou lombaires, des tassements vertébraux et des fractures provoquées par des traumatismes minimes. Cette atteinte est d’origine multifactorielle et la malabsorption de la vitamine D n’en est que l’un des éléments (27). Les troubles cardiovasculaires comprennent des réponses anormales à l’hypotension. Il existe également une hypersensibilité rénale à l’hypoxie et à l’hypotension. La fatigue est un symptôme bien repéré, mais très mal expliqué des cholestases chroniques (28). L’hypercholestérolémie et les xanthomes cutanés sont des conséquences spécifiques de la cholestase chronique, mais elles ne sont pas associées à l’athérome (29).
Abréviations
- ABCB4 : ATP binding cassette B4
- ABCC2 : ATP binding cassette C2
ou multidrug resistance associated
protein 2 (MRP2). - ABCC3 : ATP binding cassette C3
- ABCB11 : ATP binding cassette B11
- ABCC7 : ATP binding cassette C7
- ABCG5/8 : ATP binding cassette G5
ou G8
Références
- Valla DC. Cholestase. Hepato-Gastro et Oncologie Digestive 2013;20(8):618-627.
- Halilbasic E, Claudel T, Trauner M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol 2013;58(1):155-68.
- Hofmann AF. Bile acids: trying to understand their chemistry and biology with the hope of helping patients. Hepatology 2009;49(5):1403-18.
- Paumgartner G. Biliary physiology and disease: reflections of a physician-scientist. Hepatology 2010;51(4):1095-106.
- Beuers U, Hohenester S, de Buy Wenniger LJ, Kremer AE, Jansen PL, Elferink RP. The biliary HCO(3)(-) umbrella: a unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology 2010;52(4):1489-96.
- Pauli-Magnus C, Stieger B, Meier Y, Kullak-Ublick GA, Meier PJ. Enterohepatic transport of bile salts and genetics of cholestasis. J Hepatol 2005;43(2):342-57.
- Aad G, Abbott B, Abdallah J, Abdel Khalek S, Abdelalim AA, Abdesselam A, et al. Observation of spin correlation in tt events from pp collisions at radicals=7 TeV using the ATLAS detector. Phys Rev Lett 2012;108(21):212001.
- Oude Elferink RP, Paulusma CC, Groen AK. Hepatocanalicular transport defects: pathophysiologic mechanisms of rare diseases. Gastroenterology 2006;130(3):908-25.
- Fevery J. Bilirubin in clinical practice: a review. Liver Int 2008;28(5):592-605.
- Dhumeaux D, Erlinger S. Hereditary conjugated hyperbilirubinaemia: 37 years later. J Hepatol 2013;58(2):388-90.
- Jansen PL. Genetic diseases of bilirubin metabolism: the inherited unconjugated hyperbilirubinemias. J Hepatol 1996;25(3):398-404.
- Carey MC, Small DM. The physical chemistry of cholesterol solubility in bile. Relationship to gallstone formation and dissolution in man. J Clin Invest 1978;61(4):998-1026.
- Poupon R, Rosmorduc O, Boelle PY, Chretien Y, Corpechot C, Chazouilleres O, et al. Genotype- phenotype relationships in the low-phospholipid-associated cholelithiasis syndrome: A study of 156 consecutive patients. Hepatology 2013.
- Jansen PL, Muller M, Sturm E. Genes and cholestasis. Hepatology 2001;34(6):1067-74.
- Pawlikowska L, Strautnieks S, Jankowska I, Czubkowski P, Emerick K, Antoniou A, et al. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J Hepatol 2010;53(1):170-8.
- Imam MH, Gossard AA, Sinakos E, Lindor KD. Pathogenesis and management of pruritus in cholestatic liver disease. J Gastroenterol Hepatol 2012;27(7):1150-8.
- Kremer AE, van Dijk R, Leckie P, Schaap FG, Kuiper EM, Mettang T, et al. Serum autotaxin is increased in pruritus of cholestasis, but not of other origin, and responds to therapeutic interventions. Hepatology 2012;56(4):1391-400.
- Watanapa P. Recovery patterns of liver function after complete and partial surgical biliary decompression. Am J Surg 1996;171(2):230-4.
- Bulle F, Mavier P, Zafrani ES, Preaux AM, Lescs MC, Siegrist S, et al. Mechanism of gamma- glutamyl transpeptidase release in serum during intrahepatic and extrahepatic cholestasis in the rat: a histochemical, biochemical and molecular approach. Hepatology 1990;11(4):545-50.
- Eaton JE, Talwalkar JA, Lazaridis KN, Gores GJ, Lindor KD. Pathogenesis of Primary Sclerosing Cholangitis and Advances in Diagnosis and Management. Gastroenterology 2013.
- Poupon R. Primary biliary cirrhosis: a 2010 update. J Hepatol 2010;52(5):745-58.
- Padda MS, Sanchez M, Akhtar AJ, Boyer JL. Drug-induced cholestasis. Hepatology 2011;53(4):1377-87.
- Hay JE. Liver disease in pregnancy. Hepatology 2008;47(3):1067-76.
- Chand N, Sanyal AJ. Sepsis-induced cholestasis. Hepatology 2007;45(1):230-41.
- Hammel P, Couvelard A, O’Toole D, Ratouis A, Sauvanet A, Flejou JF, et al. Regression of liver fibrosis after biliary drainage in patients with chronic pancreatitis and stenosis of the common bile duct. N Engl J Med 2001;344(6):418-23.
- Corpechot C, Carrat F, Bonnand AM, Poupon RE, Poupon R. The effect of ursodeoxycholic acid therapy on liver fibrosis progression in primary biliary cirrhosis. Hepatology 2000;32(6):1196-9.
- Angulo P, Therneau TM, Jorgensen A, DeSotel CK, Egan KS, Dickson ER, et al. Bone disease in patients with primary sclerosing cholangitis: prevalence, severity and prediction of progression. J Hepatol 1998;29(5):729-35.
- Jopson L, Jones DE. Fatigue in Primary Biliary Cirrhosis: Prevalence, Pathogenesis and Management. Dig Dis 2015;33 Suppl 2:109-14.
- Sorokin A, Brown JL, Thompson PD. Primary biliary cirrhosis, hyperlipidemia, and atherosclerotic risk: a systematic review. Atherosclerosis 2007;194(2):293-9.