LIEN D’INTÉRÊTS
Aucun lien d’intérêt à déclarer
MOTS-CLÉS
Entéropathie autoimmune ; études immunohistochimiques ; lymphoprolifération intestinale
ABRÉVIATIONS
AIE : autoimmune enteropathy ; BK : bacille de Koch ; CTLA-4 : cytotoxic T-lymphocyte-associated protein 4 ; C1s : complement component 1s ; ICOS : Inducible T-cell COStimulator ; JAK : Janus tyrosine kinase ; HLA : human leucocyte antigen : LRBA :LPS-responsive beige-like anchor ; NFkB : nuclear factor-kappa B : NK : natural killer ; PAS : periodic acid schiff ; PCR : Polymerase Chain Reaction ; PD-1 : Programmed cell death 1 receptor ; TNF : tumor necrosis factor ; TNFAiP3 : TNF Alpha Induced Protein 3 ; TNFRS13B : TNF receptor superfamily member 13b
Introduction
À côté de la maladie cœliaque qui reste le diagnostic le plus fréquent d’entéropathie avec atrophie villositaire, il ne faut pas méconnaître les autres diagnostics en particulier en cas de maladie cœliaque séronégative ou de résistance au régime sans gluten. Dans certains cas, le contexte rend le diagnostic évident comme la prise médicamenteuse d’olmesartan. Dans les autres cas, il faut revoir les critères diagnostiques initiaux comme la sérologie cœliaque avant la mise sous régime sans gluten, déterminer l’haplotype HLA de type II, demander les antécédents personnels et familiaux de maladies autoimmunes et/ou de déficits immunitaires et faire réaliser au minimum une relecture anatomopathologique des biopsies initiales. Il est souvent nécessaire de renouveler les explorations endoscopiques avec études immunohistochimiques complémentaires et études de clonalité.
Les causes d’atrophie villositaire duodénale non cœliaque
Les principales causes d’atrophie villositaire et leurs critères diagnostiques sont résumés dans le tableau.
| Groupe diagnostique | Examens cliniques, paracliniques | Caractéristiques lésionnelles (en plus de l’atrophie villositaire) |
|---|
| Maladie coeliaque | IgA anti-transglutaminase ou IgG si déficit en IgA IgA anti-endomysium HLA-DQ2/DQ8 | Hyperplasie des cryptes et augmentation des lymphocytes intraépithéliaux ; richesse lymphoplasmocytaire du chorion |
| Infections Whipple Lambliase Sprue tropicale Pullulation microbienne Tuberculose intestinale VIH | PCR Whipple (biopsies duodénales et selles) Contexte géographique Breath Test Quantiféron, PCR BK sur biopsies duodénales Sérologie VIH | Macrophages PAS + G. Lamblia Granulomes avec nécrose caséeuse |
| Médicaments, causes iatrogènes Check Point Inhibitors Sartans Mycophénolate Mofétil AINS Chimiothérapie Radiothérapie GVHD | Interrogatoire Contexte clinique | Lésions GVHD |
| Inflammation/Autoimmunité Maladie de Crohn Entéropathie autoimmune +/- Déficit primitif en Ig Gastroentérite à éosinophiles | ASCA + AC anti-entérocytes+, AC anti-AIE 75KD+ Déficit en IgA, IgG et/ou IgM Anasarque, polysérite | Granulomes, distorsion glandulaire, cryptites Apoptose glandulaire, cryptites Raréfaction plasmocytaire du chorion, hyperplasie folliculaire lymphoïde Infiltrat muqueux+/- musculaire et séreux à polynucléaires éosinophiles |
| Lymphoproliférations Sprue réfractaire, EATL (complications maladie cœliaque) MEITL Lymphoproliférations T CD4+ | IgA et IgG anti-transglutaminase + HLA-DQ2/DQ8 | CD103+ CD56- Clone T CD103-CD56+ Clone T CD4+CD8-CD3+ Clone T |
Tableau : Principaux diagnostics d’atrophie villositaire
Causes infectieuses
Lambliase
Giardia lamblia (ou duodenalis) est un parasite cosmopolite fréquent transmis par voie oro-fécale via l’ingestion de kystes. La prévalence de l’infection par G. lamblia est évaluée entre 2 % et 5 % dans les pays développés et entre 4 et 42 % dans les pays en voie de développement (1). Les groupes à risque incluent les enfants et les personnes immunodéprimées comme les patients avec déficit primitif en immunoglobuline IgA (2). La sévérité de l’atrophie villositaire semble en relation avec la sévérité des symptômes (3). Dans une étude histopathologique de 30 cas de lambliase, des lésions d’atrophie villositaire étaient retrouvées dans 61 % des cas, une augmentation des lymphocytes intraépithéliaux chez 63 % des patients et 43 % avaient des lésions d’hyperplasie nodulaire folliculaire lymphoïde (4).
Maladie de Whipple
La maladie de Whipple est une cause infectieuse classique à évoquer bien que rare avec une incidence annuelle avoisinant 3/1 000 000 (5). Elle est liée à l’infection par Tropheryma whipplei, bactérie à gram positif intracellulaire. La transmission est oro-fécale, oro-orale, voire respiratoire. La bactérie se réplique par internalisation dans les macrophages puis est libérée dans les tissus par apoptose. En microscopie optique la présence d’un infiltrat macrophagique spumeux PAS positif est caractéristique. L’infection comprend 3 stades évolutifs : la phase précoce inflammatoire avec fièvre et arthralgies, puis une diarrhée avec amaigrissement et enfin des symptômes neurologiques, oculaires, rhumatologiques et cardiaques à la phase tardive (5). La présentation classique de la maladie de Whipple comprenant fièvre, diarrhée, douleurs abdominales et arthralgies, reste rare (6).
Sprue Tropicale
La sprue tropicale, fléau majeur affectant les pays en zone tropicale a une incidence de à 0,24 pour 100 000 personnes par an (7,8). Elle concerne les migrants occidentaux résidant en général depuis au moins un an en zone tropicale et les autochtones eux-mêmes. Elle a été considérée comme une cause majeure de malabsorption et de décès en Inde dans les années soixante (9). Les principales autres régions tropicales concernées sont l’Asie du Sud-Est, l’Afrique, les Philippines et certaines îles des Caraïbes comme Haïti où la prévalence de la sprue tropicale est estimée à 43 %, Cuba et Porto Rico où la prévalence est de 11 % (10,11,12). De façon assez surprenante, l’incidence de la sprue tropicale a considérablement diminué jusqu’à presque disparaître chez les expatriés vivant en zones d’endémie, probablement en raison du recours plus systématique aux antibiotiques en cas de diarrhée et aux meilleures pratiques sanitaires (13,14,15). La description initiale de la sprue tropicale fait état de deux phases : une phase d’installation brutale avec un tableau de gastroentérite, puis une phase chronique avec diarrhée et syndrome de malabsorption. Typiquement, les patients présentent en phase chronique un amaigrissement, une asthénie, une anémie macrocytaire liée à une carence en folates et vitamine B12, une hypoalbuminémie et une stéatorrhée. Les coprocultures et examens parasitologiques des selles sont négatifs à ce stade (16). Chez les enfants des zones d’endémie, la diarrhée chronique et la malabsorption peuvent induire un retard de croissance (17).
Dans la sprue tropicale, l’analyse histopathologique du grêle montre une atrophie villositaire, généralement de type partiel. Tout le grêle peut être concerné mais l’atrophie villositaire se retrouve classiquement au niveau de l’iléon terminal contrairement à la maladie cœliaque qui prédomine dans le grêle proximal (8,18). La physiopathogénie de la sprue tropicale reste totalement méconnue. Le facteur environnemental semble jouer un rôle prépondérant tel qu’en témoignent les cas de soldats américains initialement totalement asymptomatiques qui ont développé une sprue tropicale après plusieurs mois passés au Vietnam et qui ont totalement guéri après le retour aux USA (19). Un cercle physiopathogénique a été proposé impliquant une hyperproduction d’entéroglucagon suite à l’infection intestinale aiguë et à la destruction épithéliale intestinale. L’hypersécrétion d’entéroglucagon entraînerait le ralentissement du transit et une pullulation microbienne responsable du syndrome de malabsorption (10).
Pullulation microbienne
Le syndrome de pullulation microbienne du grêle observé dans nos pays occidentaux est généralement secondaire à des modifications anatomiques (sténose, anse borgne, diverticulose grêlique) ou fonctionnelles (achlorhydrie, réduction de la motilité intestinale) du grêle et peut s’accompagner d’une malabsorption des graisses dans les formes les plus sévères (20). Alors que plus de la moitié des patients ont une histologie duodénale normale, environ 24 % d’entre eux présentent des lésions d’atrophie villositaire (21).
Infections virales
Des lésions d’atrophie villositaire intestinale sont retrouvées jusqu’à 54 % des personnes infectées par le VIH sans infection opportuniste (22). Ces lésions pourraient être liées aux désordres immunitaires induits par le virus lui-même puisqu’avant la déplétion en lymphocytes T CD4+, l’infection aiguë par le VIH induit une infiltration muqueuse par des lymphocytes T effecteurs mémoires CD4+ et CD8+ T à l’origine d’une destruction de l’épithélium intestinal par apoptose (23). En histologie, on observe une atrophie villositaire généralement partielle avec une hyperplasie des cryptes (22).
Chez les immunodéprimés, d’autres virus peuvent être impliqués. C’est le cas des patients transplantés rénaux sous immunosuppresseurs, qui développent des lésions d’atrophie villositaire partielle au cours d’infections chroniques par Norovirus par défaut d’élimination du pathogène (24,25).
Causes inflammatoires et auto-immunes
Maladie de Crohn
Des lésions d’atrophie villositaire ont été décrites au cours de la maladie de Crohn (26). Il existe également une association entre maladie de Crohn et maladie cœliaque ; pour confirmer le diagnostic de maladie cœliaque, il est nécessaire de réévaluer les lésions d’atrophie villositaire après au moins 1 an de régime sans gluten (27).
Gastroentérite à éosinophiles
La gastroentérite à éosinophiles est caractérisée par l’existence d’une symptomatologie gastro-intestinale (douleurs abdominales, dyspepsie, diarrhée, nausées et vomissements), la présence d’une infiltration à éosinophiles limitée à la paroi digestive et l’absence de facteur causal tels qu’une cause médicamenteuse (sels d’or, clozapine, carbamazepine, rifampicine, gabapentine et prégabaline, etc.), un syndrome hyperéosinophilique, une maladie de Crohn ou autre maladie de système ainsi qu’une pathologie parasitaire intestinale. Klein et al. a proposé une classification en fonction de l’infiltration éosinophilique de la paroi digestive permettant de distinguer les infiltrations à prédominance muqueuse (jusqu’à 100 % des cas), musculeuse (jusqu’à 70 % des cas) et séreuse (jusqu’à 13 % des cas) (28,29). Le côlon et l’œsophage peuvent être atteints en plus de l’estomac et du grêle (30,31,32,33). Plusieurs chimiokines et médiateurs inflammatoires comme l’interleukine 5 participent au recrutement des polynucléaires éosinophiles dans le tractus gastro-intestinal (30). Lorsque la musculeuse est atteinte, un syndrome obstructif intestinal peut être observé, voire une ascite lorsque l’extension se fait jusqu’à la séreuse. Des cas de péritonite sont possibles. Une hyperéosinophilie sanguine peut être observée de façon non obligatoire. L’aspect endoscopique est non spécifique et inclut ulcérations, érythème et pseudopolypes (34).
Entéropathie auto-immune
L’entéropathie auto-immune reste une cause rare et peu connue de diarrhée chronique. Les critères diagnostiques retenus par la Mayo Clinic sont : une diarrhée chronique évoluant depuis plus de 6 mois, un syndrome de malabsorption, des signes histologiques spécifiques incluant une atrophie villositaire et une hyperlymphocytose intraépithéliale modérée et l’exclusion des autres causes d’atrophie villositaire (35). De plus, les symptômes cliniques et l’atrophie villositaire intestinale sont résistants à tout régime d’exclusion alimentaire. Si l’entéropathie autoimmune a d’abord été majoritairement décrite chez l’enfant avec le syndrome d’IPEX pédiatrique (Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked), secondaire à une mutation du gène FOXP36) 3), les descriptions chez l’adulte sont en constante augmentation (35,37,38). Une des premières cohortes adultes a été décrite à la Mayo Clinic en 2007. Elle comprenait 15 patients avec diarrhée chronique et syndrome de malabsorption résistant au régime sans gluten (35). Les lésions histologiques étaient une atrophie villositaire intestinale avec une augmentation modérée des lymphocytes intraépithéliaux. Il pouvait s’y associer des lésions d’apoptose glandulaire comparables à celles observées lors de la réaction du greffon contre l’hôte ainsi que des lésions de cryptite et d’abcès cryptiques. Les autres atteintes digestives étaient des gastrites lymphocytaires et des colites lymphocytaires observées chez 80 % des patients. Il est donc nécessaire d’associer une coloscopie à l’endoscopie digestive haute et de réaliser des biopsies gastriques, duodénales et coliques étagées pour le bilan d’une entéropathie auto-immune. Il est parfois difficile de différencier une entéropathie auto-immune d’une maladie cœliaque réfractaire au régime sans gluten. Dans la série de la Mayo Clinic, la majorité des patients (14/13) avaient des anticorps anti-entérocyte et un tiers des patients avaient des anticorps anti-transglutaminase (15/5) Si les anticorps anti-transglutaminase peuvent être observés au cours d’une entéropathie auto-immune, les anticorps anti-entérocyte semblent en revanche spécifiques des entéropathies auto-immunes lorsqu’on ne retient comme tests positifs que les forts niveaux de fluorescence (39). Il est également possible de détecter des anticorps dirigés contre une protéine présente dans la bordure en brosse des entérocytes de 75 kDa par une méthode radio-immunologique qui permet un dosage spécifique et précis du taux d’anticorps anti-AIE75kDa (40). L’absence d’anticorps anti-entérocyte ne suffit pas à exclure le diagnostic d’entéropathie autoimmune. Récemment des outils génétiques ont été développés pour aider au diagnostic d’entéropathie en particulier lorsque les patients résistants au régime sans gluten présentent des IgA anti-transglutaminase et l’haplotype HLADQ2/8 de susceptibilité à la maladie cœliaque. Le laboratoire d’immunité intestinale de l’institut Imagine (Inserm UMR1163) a développé un panel de séquençage ciblé « Entéropathie » permettant une étude génétique constitutionnelle des patients avec entéropathie. Nos travaux montrent qu’un variant génétique pathogénique est retrouvé chez environ 40 % des patients avec entéropathie autoimmune. Les gènes concernés sont : STAT1, STAT3, LRBA, CTLA4, ICOS, TNFRS13B, TNFAIP3, NFKB1, C1S (41).
Les entéropathies auto-immunes peuvent être observées chez les patients avec déficit primitif en immunoglobulines. Deux des 15 patients de la cohorte décrite par Akram et al. avaient un déficit immunitaire commun variable (35). Plus récemment une étude italienne décrivant 40 patients avec entéropathie auto-immune note un déficit immunitaire chez 21 d’entre eux, en particulier un déficit immunitaire commun variable (DICV) et une agammaglobulinémie liée à l’X (42). La forte proportion de patients avec hypogammaglobulinémie primitive explique le faible taux de positivité des anticorps anti-entérocyte de 14 % (4/28) dans cette série. Jusqu’à 50 % des patients atteints de DICV se plaignent d’une diarrhée chronique. Après élimination des causes infectieuses et d’une hypogammaglobulinémie secondaire à une entéropathie exsudative, le diagnostic d’entéropathie auto-immune associée au DICV doit être évoqué. On trouve des lésions d’atrophie villositaire intestinale chez la moitié des patients DICV avec symptômes digestifs et une hyperlymphocytose intraépithéliale intestinale chez les trois quarts d’entre eux (43). Néanmoins, atrophie villositaire et hyperlymphocytose intraépithéliale sont généralement moins sévères que dans la maladie cœliaque et les plasmocytes sont souvent rares ou absents (43). Des lésions de colite peuvent être associées, souvent de type microscopique (plus souvent lymphocytaire que collagène), ou de maladie inflammatoire chronique intestinale avec distorsion de l’architecture glandulaire et ulcérations. À ces lésions peuvent être intriquées des lésions d’hyperplasie folliculaire lymphoïde diffuse et d’apoptose glandulaire de type greffon contre l’hôte (GVHD : Graft Versus Host Disease) (43). Leur présence aide à éliminer le diagnostic de maladie cœliaque, les anticorps anti-transglutaminase étant négatifs du fait du déficit en production d’anticorps et l’haplotype HLA-DQ2/DQ8 de susceptibilité de la maladie cœliaque peu discriminant car observé chez une grande majorité (jusqu’à 77 %) des patients DICV avec manifestations digestives (43). L’habituelle absence de réponse au régime sans gluten permet d’éliminer définitivement le diagnostic de maladie cœliaque (43).
Causes médicamenteuses / iatrogènes
Check point inhibitors
Des lésions d’atrophie villositaire avec infiltration lymphoplasmocytaire du chorion et hyperlymphocytose intraépithéliale ont été observées avec l’utilisation des anticorps anti-PD-1 utilisés dans le cadre d’une immunothérapie anti-tumorale (44).
Sartans
L’olmesartan est responsable de la majorité des entéropathies d’origine médicamenteuse (45). Les autres inhibiteurs du récepteur de l’angiotensine sont beaucoup moins impliqués. L’entéropathie à l’olmésartan, inhibiteur du récepteur de l’angiotensine 2 peut mimer une maladie cœliaque en raison de l’hyperlymphocytose intraépithéliale et des lésions d’atrophie villositaire. Toutefois certaines caractéristiques histologiques (lésions d’apoptose glandulaire) et sérologiques (négativité des anticorps cœliaques, positivité des anticorps anti-entérocyte ou anti-AIE 75KD observée chez 40 % des patients) la rapprochent davantage des entéropathies auto-immunes (46). L’âge (en moyenne 70 ans) et la notion de prise d’olmésartan sont des éléments diagnostiques clés chez des patients présentant brusquement une diarrhée profuse et un syndrome de malabsorption. Il existe fréquemment un terrain auto-immun et les patients sont porteurs de l’haplotype de susceptibilité de la maladie cœliaque HLA DQ2-DQ8 dans environ 68 % des cas (47). Dans notre expérience l’hyperlymphocytose intraépithéliale duodénale était modérée (30-40 %) à l’exception d’un patient avec hyperlymphocytose élevée (100 %) qui avait également une gastrite lymphocytaire (46). Les sartans sont également pourvoyeurs de sprue collagène avec un épaississement de la membrane basale collagène sous épithéliale (46).
Mycophenolate Mofetil
Le Mycophenolate mofetil est la cause la plus courante d’atrophie villositaire chez les transplantés d’organes (48). L’entéropathie se développe typiquement dans l’année suivant son introduction et est responsable de diarrhée chronique (48). Les lésions sont liées à la fois à la toxicité directe ainsi qu’à l’inhibition de la prolifération cellulaire causée par l’inhibition de la synthèse des purines.
Lymphoproliférations intestinales
L’intestin est une localisation extra-ganglionnaire fréquente des lymphomes périphériques (49,50). Les lymphomes intestinaux avec atrophie villositaire les plus fréquents sont l’enteropathy-associated T-cell lymphoma (EATL) de type I, EATL de type 2, les lymphoproliférations T et NK indolentes et le lymphome du MALT (mucosa-associated lymphoid tissue). Bien que ne représentant que 5 % des lymphomes T périphériques, l’EATL de type I est le plus fréquent des lymphomes intestinaux primitif dans les pays occidentaux et est associé à la maladie cœliaque (49). Des ulcérations du grêle peuvent être à l’origine d’hémorragie digestive et les sténoses d’un syndrome occlusif. Il révèle la maladie cœliaque dans 40 % des cas. Dans les autres cas, la maladie cœliaque est connue, voire compliquée d’une sprue réfractaire clonale (sprue de type II). L’EATL de type II, encore appelé MEITL (monomorphic epitheliotropic intestinal T-cell lymphoma), est plus fréquent en Asie chez l’homme et survient généralement au cours de la cinquantaine. II est caractérisé par une prolifération monomorphe de lymphocytes de taille petite à moyenne CD3+, CD8+/-, TCRab+/-, CD57-, CD4-, CD5-, TiA1+, présentant des réarrangements du TCR et exprimant CD56. Peut-être faut-il rapprocher ces lymphomes des rares cas de lymphome malin non hodgkinien (LMNH) de type NK/T observés dans l’intestin. Contrairement à l’EATL de type I, il n’existe aucun lien avec la maladie cœliaque. L’infiltration tumorale, volontiers étendue à l’ensemble du tractus gastro-intestinal, confère un aspect d’atrophie villositaire avec hyperlymphocytose intraépithéliale. Son mode de révélation peut être une diarrhée chronique avec amaigrissement, voire une perforation intestinale (51,52,53).
Les lymphoproliférations indolentes de l’intestin sont des proliférations clonales T (CD4+, CD8+, CD4-CD8-), ou de type Natural Killer. Elles n’ont généralement pas de lien avec la maladie cœliaque et peuvent s’étendre à tout le tractus gastro-intestinal, même si l’atteinte grêlo-colique est majoritaire. Même indolentes, ces lymphoproliférations induisent douleurs abdominales, diarrhée chronique et malnutrition (54,55). Les lymphoproliférations T CD4+ du chorion correspondent à des proliférations clonales à petites cellules CD4+ du chorion intestinal (54,55) qui peuvent envahir l’épithélium causant un enrichissement épithélial en lymphocytes T CD4+ entraînant une baisse relative des LIE CD8+ et conférant ainsi un aspect de trou phénotypique pouvant faire évoquer initialement le diagnostic de sprue réfractaire. Il n’existe pas d’aspect macroscopique spécifique avec en endoscopie la possibilité de lésions nodulaires, polypoïdes, ulcérées ou simplement atrophiques. En histologie l’atrophie, villositaire est habituellement partielle (55).
Démarche diagnostique
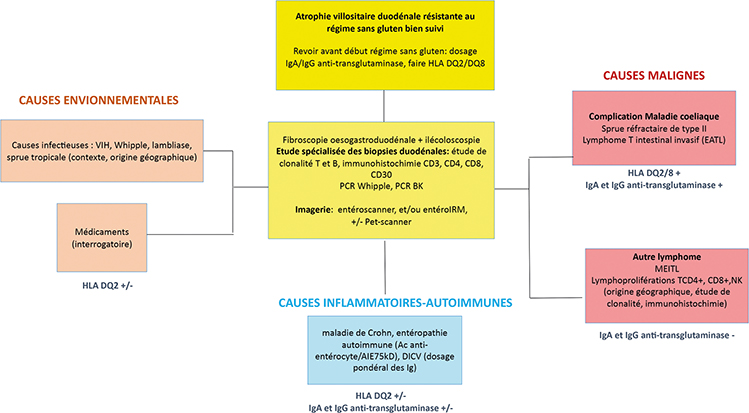
La démarche diagnostique d’une entéropathie avec atrophie villositaire résistante au régime sans gluten est résumée dans la figure.
Bilan endoscopique et réalisation des biopsies digestives
L’atrophie villositaire duodénale résistante au régime sans gluten nécessite au minimum une relecture anatomopathologique et en général de renouveler le bilan endoscopique (fibroscopie œsogastro-duodénale, iléocoloscopie avec biopsies étagées gastriques, duodénales et iléocoliques). Un examen complémentaire par vidéocapsule endoscopique peut être utile pour apprécier l’extension de l’atteinte ainsi que l’évaluation d’autres lésions associées comme des ulcérations aphtoïdes. Elle est notablement utile au diagnostic de maladie de Crohn grêlique lorsque la coloscopie est sans anomalie, en particulier en cas d’élévation importante de la calprotectine fécale (56).

Figure
L’aspect macroscopique des différentes maladies gastro-intestinales avec atrophie villositaire est généralement non spécifique. Il peut exister un aspect en mosaïque, crénelé, fissuraire de la muqueuse duodénale témoignant de l’atrophie villositaire mais également des ulcérations comme dans la maladie de Crohn, la gastroentérite à éosinophiles, certaines entéropathies autoimmunes et lymphoproliférations intestinales. Comme dans la maladie cœliaque, les lésions d’atrophie villositaire peuvent être hétérogènes dans les entéropathies auto-immunes et à éosinophiles notamment (57) : il est donc nécessaire de réaliser 2 biopsies dans le bulbe et 6 au minimum dans la deuxième partie du duodénum.
Il ne faut pas hésiter à réaliser des biopsies congelées pour étude de clonalité T et B, même si des études de clonalité peuvent également être réalisées sur biopsies en paraffine. En effet, l’étude TNGS (Targeted Next Generation Sequencing) se réalise sur biopsies congelées, étude qui permet dans un second temps de guider la prise en charge thérapeutique des lymphoproliférations intestinales grâce à l’identification de la ou des mutations somatiques retrouvées.
Des biopsies duodénales doivent également être conditionnées pour recherche microbiologique de pathogènes spécifiques comme les PCR Whipple et PCR BK.
Si l’atrophie villositaire et l’hyperplasie des cryptes du chorion ne permettent pas en elles-mêmes d’identifier la cause de l’entéropathie, certains critères histopathologiques orientent fortement le diagnostic. C’est le cas en pathologie infectieuse de l’aspect spumeux spécifique des macrophages PAS+ au cours de la maladie de Whipple, des granulomes tuberculoïdes avec nécrose caséeuse au cours de la tuberculose digestive ou de la visualisation directe sur coupe du parasite comme au cours des infections à G. lamblia.
D’autres critères aident au diagnostic des causes inflammatoires et auto-immunes comme les granulomes sans nécrose caséeuse des maladies de Crohn et la raréfaction plasmocytaire du chorion associée aux entéropathies des patients avec déficit primitif en immunoglobulines. Un taux d’éosinophiles supérieur à 20 polynucléaires par champs permet le diagnostic de la gastroentérite à éosinophiles.
Alors que l’infiltration tumorale des lymphomes à grandes cellules comme dans l’EATL pose rarement de problème diagnostique, il existe une errance diagnostique courante pour le diagnostic des lymphoproliférations intestinales « indolentes » à petites cellules où il n’existe habituellement pas de trouble architectural marqué. Les patients souffrent alors de diarrhée, malnutrition avec hypoalbuminémie liées à une entéropathie avec atrophie villositaire résistante au régime sans gluten pendant en moyenne 7 années avant le diagnostic (55). En plus de l’étude de clonalité, les études histochimiques complémentaires sont indispensables au diagnostic. Au cours des lymphoproliférations CD4+, le marquage CD3, CD8, CD4 permet l’identification d’un excès de lymphocytes CD4+ dans le chorion qui infiltrent l’épithélium intestinal habituellement riche en lymphocytes intraépithéliaux CD3+CD8+. Il existe donc un excès de lymphocytes CD3+CD8- donnant un aspect de trou phénotypique qui peut faire, avec la présence d’un clone T identifié par PCR, poser en première intention le diagnostic de sprue cœliaque réfractaire clonale (de type II). La différence majeure réside sur la positivité CD4+ de ces lymphocytes intraépithéliaux CD3+CD8- au cours de ces lymphoproliférations, qui sont négatifs pour le CD4 au cours de la sprue réfractaire de type II. En plus de l’étude immunohistochimique sur coupes, l’étude phénotypique en cytométrie de flux des lymphocytes isolés de l’intestin est très utile pour préciser le type de lymphoproliférations à petites cellules (55).
Bilan complémentaire
La recherche d’une cause infectieuse implique des examens complémentaires spécifiques comme le test Quantiféron pour la tuberculose, la PCR Whipple dans les selles ou le Breath test au glucose (ou lactulose) pour la pullulation microbienne. Ce dernier peut également être utilisé pour la sprue tropicale étant donné qu’au stade de malabsorption les coprocultures et examens parasitologiques des selles sont habituellement négatifs (16). Le dosage des anticorps anti-entérocyte ou anti-AIE 75kDa est utile au diagnostic d’entéropathie auto-immune. L’absence de ces anticorps anti- entérocyte ne permet toutefois pas d’écarter le diagnostic, principalement en raison de l’association possible à un déficit immunitaire primitif.
Alors que le diagnostic du déficit immunitaire acquis (sérologie VIH ou contexte de prise au long cours d’immunosuppresseurs) est assez simple, le déficit immunitaire primitif peut être de diagnostic plus difficile chez l’adulte. La réalisation d’une électrophorèse des protéines sériques estrecommandée pour dépister un déficit primitif en immunoglobulines (Ig) en montrant typiquement une hypogammaglobulinémie d’intensité variable concomitante d’un taux d’albumine normal écartant ainsi l’hypothèse d’une hypogammaglobulinémie secondaire à une entéropathie exsudative. Le bilan comprend aussi une numération et un immunophénotypage des sous populations lymphocytaires T, B et NK, l’évaluation des réponses vaccinales par tests fonctionnels appropriés ainsi qu’un dosage en sous classes d’Ig (IgA, IgG, IgM et sous classes d’IgG). Après le déficit sélectif en immunoglobuline IgA, le déficit immunitaire commun variable (DICV) est le plus fréquent des déficits de l’immunité humorale et est caractérisé par une diminution d’au moins deux déviations standards de deux à trois sous-classes d’Ig. Sa prévalence est estimée à 1/50 000 à 1/25 000 en population caucasienne (58). Le diagnostic est également évoqué devant des infections ORL et broncho-pulmonaires à répétition pouvant conduire à une dilatation des bronches (58).
Les examens d’imagerie du grêle (Entéro-scanner, entéro-IRM) sont utiles au diagnostic et nécessaires avant la réalisation d’une vidéocapsule endoscopique en raison des risques de sténoses en cas de pathologie crohnienne ou lymphomateuse. Le Pet-Scanner est utile pour distinguer les entéropathies inflammatoires/auto-immunes des lymphoproliférations intestinales en particulier en cas de lymphomes invasifs pourvoyeurs d’intenses foyers de fixation (EATL, MEITL etc.) (55).
Traitement, évolution et risques évolutifs
Le traitement de l’entérite infectieuse repose sur le traitement de l’agent infectieux responsable. Le traitement des lambliases repose sur les imidazolés, flagyl et fasigyne dont les cures peuvent être répétées chez l’immunodéprimé (55,59).
Le traitement de la maladie de Whipple nécessite une antibiothérapie au long cours. Le plus souvent elle associe un traitement « d’attaque », par exemple pénicilline G et streptomycine injectées pendant 2 semaines puis cotrimoxazole (triméthoprime + sulfaméthoxazole) per os pendant un à deux ans (5). L’utilisation de l’hydroxychloroquine a été proposée en association avec la doxycycline pour prévenir les formes résistantes ou en rechute sous cotrimoxazole (60).
Le traitement de la sprue tropicale repose classiquement sur l’utilisation de cyclines, les tétracyclines étant les antibiotiques les plus couramment utilisés (61). Plus rarement, les sulfamides et les fluoroquinolones sont employés (16). La durée du traitement n’est pas codifiée pouvant s’étendre de quelques semaines à quelques mois, parfois supérieure à 6 mois pour les malades vivant en zone d’endémie ayant une maladie prolongée (16). Outre le traitement antibiotique, une supplémentation des carences en particulier en folates et vitamine B12 doit être réalisée. Le risque de récidive existe pour les malades vivant en zone tropicale ce qui nécessite un suivi prolongé (62).
Le traitement de la pullulation microbienne repose également sur un traitement antibiotique dont l’efficacité permet également de confirmer le diagnostic. Les fluoroquinolones, l’acide clavulanique-amoxicilline, le métronidazole et la rifaximine sont les plus employés. L’utilisation des probiotiques pourrait également accélérer la guérison (63,64). En effet, Il existerait jusqu’à 40 % d’échec thérapeutique et 44 % des patients pourraient avoir une pullulation microbienne récidivante en dépit de l’antibiothérapie.
Alors que la thérapie anti-rétrovirale est essentielle au traitement de l’infection par le VIH, son impact sur l’entéropathie est généralement retardée (65,66). La restauration de la population T intestinale peut être lente et incomplète. Des lésions d’atrophie villositaire persistante ont été retrouvées chez les patients VIH traités par anti-rétroviraux à long terme alors qu’ils avaient reconstitué leurs taux de lymphocytes T CD4+ dans le sang veineux périphérique (65). Le traitement des entéropathies médicamenteuses repose sur l’arrêt du médicament responsable. Toutefois il est souvent nécessaire de recourir aux corticoïdes généraux, voire à l’administration d’anticorps anti-TNF-alpha dans les entérocolites induites par les immunothérapies anti-tumorales. Lorsque l’atteinte du grêle est isolée, on peut envisager le recours au budésonide qui peut également être utilisé pour raccourcir l’évolution des entéropathies aux sartans (46). Dans le cas des transplantés d’organe, il n’est parfois pas possible d’arrêter le mycophenolate mofetil ; la réduction de doses suffit dans certains cas à stopper la diarrhée (48).
Le traitement de causes inflammatoires et auto-immunes d’atrophie villositaire repose sur les corticoïdes et les immunosuppresseurs. C’est le cas notamment de la gastroentérite à éosinophiles pour laquelle les anti-histaminiques peuvent également être utilisés. Le budésonide « open capsule » (le patient avale le contenu de la gélule en le mâchant préalablement pour permettre l’action du budésonide dans le grêle proximal) est le traitement de première intention de l’entéropathie auto-immune (38). En cas de déficit en Ig associé, la supplémentation en Ig n’a d’effet ni sur la fréquence des infections gastro-intestinales ni sur les lésions spécifiques comme l’atrophie villositaire intestinale (43). Le recours aux immunosuppresseurs parait justifié pour les entéropathies autoimmunes cortico-résistantes ou dépendantes. Parmi eux, la ciclosporine, le tacrolimus, le sirolimus et le cyclophosphamide ont beaucoup été utilisés avant l’apparition des anticorps anti-TNF-alpha avec une efficacité variable (39,67). L’identification des causes génétiques des entéropathies auto-immunes permet le recours aux thérapies ciblées dans certains cas. Nous avons notamment rapporté les effets d’une thérapie ciblée chez une jeune femme de 25 ans suivie pour une entérocolite sévère depuis l’âge de 6 mois avec atrophie villositaire duodénale et colite microscopique dans un contexte d’hypogammaglobulinémie primitive sans amélioration après plusieurs lignes de traitement dont les corticoïdes, des immunosuppresseurs et des biothérapies. L’identification d’une mutation gain de fonction de STAT3 (Signal transducer and activator of transcription 3) a conduit à proposer un traitement par le ruxolitinib, un inhibiteur de JAK1/2 bloquant la voie de signalisation de STAT3. Ce traitement a permis une rémission clinique, biologique et histologique complète et prolongée (68). Une série récente de 17 patients avec mutation de STAT1 ou STAT3 traités par inhibiteurs de JAK rapporte une amélioration clinique significative chez 14 d’entre eux (69). Par ailleurs, l’abatacept, une protéine de fusion comprenant la partie extra cellulaire de CTLA4 et agoniste du récepteur a montré son efficacité dans le traitement des manifestations auto-immunes notamment digestives chez des patients porteurs d’une mutation perte de fonction de CTLA4 (70).
Le pronostic des EATL et des MEITL reste sombre en raison de leur faible chimiosensibilité. Comme plus de 80 % des EATL expriment le CD30 (71) un protocole français en cours inclut l’utilisation des anticorps anti-CD30 à la chimiothérapie (ClinicalTrials.gov Identifier: NCT03217643). Il n’existe pas de traitement codifié pour les lymphoproliférations indolentes. Les corticoïdes et les anticorps anti-CD52 peuvent induire une rémission clinique (55). Il existe toutefois un risque de transformation en lymphome de haut grade (55). L’identification des mutations acquises par TNGS et/ou cytogénétique peut être utile pour guider la thérapeutique (72).
Conclusion
Les causes d’atrophie villositaire intestinale sont diverses. L’interrogatoire et l’origine géographique peuvent orienter facilement vers une cause infectieuse ou médicamenteuse. Le diagnostic d’entéropathie auto-immune et de lymphoproliférations indolentes est plus difficile et nécessite la réalisation de biopsies spécialisées. Le traitement de ces deux entités reste mal codifié et implique souvent en première intention le budésonide. La réalisation d’études génétiques constitutionnelles permet maintenant d’envisager des thérapies ciblées dans les entéropathies autoimmunes et la cytogénétique ou le TNGS somatique celui des lymphoproliférations indolentes.
Références
- Caccio SM, Thompson RA, McLauchlin J, Smith HV, Unravelling Cryptosporidium and Giardia epidemiology, Trends 21(2005)430-7.
- Langford D, Housley M, Boes M, et al. Central Importance of Immunoglobulin A in Host Defense against Giardia Infection and Immunity. 2002;70(1):11-8.
- Duncombe V, Bolin T, Davis A, et al. Histopathology in giardiasis: a correlation with Aust N Z J Med. 1978;8(4):392-6.
- Arevalo F, Aragon V, Morales L, et al. Duodenal villous atrophy, an unexpectedly common finding in giardia lamblia Rev Gastroenterol Peru. 2010;30(4):272-6.
- Dolmans R, Boel C, Lacle M, et al. Clinical manifestations, treatment, and diagnosis of Tropheryma whipplei Clin Microbiol Rev. 2017;30(2):529-55.
- Hujoel I, Johnson D, Lebwohl B, et al. Tropheryma whipplei infection (Whipple disease) in the United Dig Dis Sci. 2019;64/213-23
- Booth The 1st Description of Tropical Sprue (Willian Hillary). Gut 1964;5:45-50.
- Pipaliya N, Ingle M, Rathi C, et al. Spectrum of chronic small bowel diarrhea with malabsorption in Indian subcontinent: is the trend really changing? Intest 2016;14(1):75-82.
- Mathan VI, Baker SJ. Epidemic tropical sprue and other epidemics of diarrhea in South Indian villages. The American journal of clinical nutrition 1968;21:1077-87.
- Cook Aetiology and pathogenesis of postinfective tropical malabsorption (tropical sprue). Lancet 1984;1:721-3.
- Booth Tropical sprue. Lancet 1984;1 (8384):1018.
- Klipstein Sprue and subclinical malabsorption in the tropics. Lancet 1979;1:277-8.
- Bartholomew William Hillary and sprue in the Caribbean: 230 years later. Gut 1989;30 Spec No:17-21.
- Klipstein Tropical sprue in travelers and expatriates living abroad. Gastroenterology 1981;80:590-600.
- Rosenberg Tropical enteritis: nutritional consequences and connections with the riddle of cholera. The Journal of nutrition 2003;133:333S-5S.
- Macaigne G, Boivin JF, Auriault ML, et al. Tropical sprue: two cases in the Paris Gastroenterologie clinique et biologique 2004;28:913-6.
- Farthing Tropical malabsorption. Seminars in gastrointestinal disease 2002;13:221-31.
- Langenberg M, Wismans P, van Genderen Distinguishing tropical sprue from celiac disease in returning travellers with chronic diarrhoea: a diagnostic challenge? Travel Med Infect Dis. 2014 Jul-Aug;12(4):401-5.
- Haghighi P, Wolf Tropical sprue and subclinical enteropathy: a vision for the nineties. Critical reviews in clinical laboratory sciences 1997;34:313-41.
- Thorens J, Froehlich F, Schwizer W, et al. Bacterial overgrowth during treatment with omeprazole compared with cimetidine: a prospective randomised double blind Gut 1996;39:54-9.
- Lappinga P, Abraham S, Murray J, et al. Small Intestinal Bacterial Overgrowth: Histopathologic Features and Clinical Correlates in an Underrecognized Archives of Pathology & Laboratory Medicine. 2010;134(2):264-170.
- Sakai E, Higurashi T, Ohkubo H, et al. Investigation of Small Bowel Abnormalities in HIV-Infected Patients Using Capsule Gastroenterol Res Pract. 2017;2017:1932647.
- Epple H, Allers K, Troger H, et al. Acute HIV infection induces mucosal infiltration with CD4+ and CD8+ T cells, epithelial apoptosis, and a mucosal barrier defect. Gastroenterology. 2010;139(4):1289-300.
- Westhoff TH, Vergoulidou M, Loddenkemper C, et al. Chronic norovirus infection in renal transplant Nephrol Dial Transplant 2009;24:1051-3.
- Troeger H, Loddenkemper C, Schneider T, et al. Structural and functional changes of the duodenum in human norovirus Gut 2009;58:1070-7.
- Marquès Camí M, Serracarbasa A, D’Haens G, et al. Characterization of Mucosal Lesions in Crohn’s Disease Scored With Capsule Endoscopy: A Systematic Review Front Med (Lausanne)2021 Jan 14;7:600095.
- Alkhayyat M, Abureesh M, Almomani A, et al. Patients With Inflammatory Bowel Disease on Treatment Have Lower Rates of Celiac Inflamm Bowel Dis 2021 14;izab084.
- Talley N, Shorter R, Phillips S, Zinsmeister Eosinophilic gastroenteritis: a clinicopathological study of patients with disease of the mucosa, muscle layer, and subserosal tissues. Gut. 1990;31(1):54-8.
- Klein N, Hargrove R, Sleisenger M, et al. Eosinophilic Medicine (Baltimore). 1970;49(4):299-319.
- Jensen E, Martin C, Kappelman M, et al. Prevalence of eosinophilic gastritis, gastroenteritis, and colitis: Estimates from a national administrative J Pediatr Gastroenterol Nutr. 2016;62(1):36-42.
- Chang J, Choung R, Lee R, et al. A shift in the clinical spectrum of eosinophliic gastroenteritis toward the mucosal disease Clin Gastroenterol Hepatol. 2010;8(8):669-75.
- Reed C, Woosley J, Dellon Clinical characteristics, treatment outcomes, and resource utilization in children and adults with eosinophilic gastroenteritis. Dig Liver Dis. 2015;47(3):197-201.
- Butterfield J, Murray Eosinophilic gastroenteritis and gluten-sensitive enteropathy in the same patient. J Clin Gastroenterol. 2002;34(5):552-3.
- Tien F, Wu J, Jeng Y, et al. Clinical features and treatment responses of children with eosinophilic Pediatr Neonatol. 2011;52(5):272-8.
- Akram S, Murray JA, Pardi DS, et al. Adult autoimmune enteropathy: Mayo Clinic Rochester Clin Gastroenterol Hepatol Off Clin Pract J Am Gastroenterol Assoc 2007;5 (11):1282-90
- Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 2001;27(1):20-1.
- Corazza GR, Biagi F, Volta U, et al. Autoimmune enteropathy and villous atrophy in Lancet Lond Engl 1997;350(9071):106-9.
- Elli L, Ferretti F, Vaira Demystifying autoimmune small bowel enteropathy. Curr Opin Gastroenterol 2019;35 (3):243-9.
- Di Sabatino A, Biagi F, Lenzi M, et al. Clinical usefulness of serum antibodies as biomarkers of gastrointestinal and liver Dig Liv Dis 2017; 49: 947–56
- Kobayashi I, Imamura K, Kubota M, et al. Identification of an autoimmune enteropathy-related 75-kilodalton Gastroenterology 1999;117 (4):823-30.
- Haas M, Charbit-Henrion F, Chaussade S, et al. Enteropathie autoimmune : diagnostic et traitement en 2021 ; Hépato-Gastro et Oncologie Volume 28 (3) 2021. 383-9.
- Villanacci V, Lougaris V, Ravellic A, et al. Clinical manifestations and gastrointestinal pathology in 40 patients with autoimmune Clin Immunol 2017; 207: 10-7
- Malamut G, Verkarre V, Suarez F, et al. The enteropathy associated with common variable immunodeficiency: the delineated frontiers with celiac disease. Am J Gastroenterol 2010;105(10):2262-75.
- Collins M, Michot JM, Danlos FX, et al. Inflammatory gastrointestinal diseases associated with PD-1 blockade Ann Oncol 2017;28:2860–5.
- DeGaetani M, Tennyson C, Lebwohl B, et al. Villous atrophy and negative celiac serology: a diagnostic and therapeutic Am J Gastroenterol. 2013 May;108(5):647-53.
- Scialom S, Malamut G, Meresse B, et al. Gastrointestinal Disorder Associated with Olmesartan Mimics Autoimmune PLoS ONE. 2015;10(6):e0125024.
- Rubio-Tapia A, Herman M, Ludvigsson J, et al. Severe spruelike enteropathy associated with Mayo Clin Proc. 2012 Aug;87(8):732-8.
- Weclawiak H, Ould-Mohamed A, Bournet B, et al. Duodenal villous atrophy: a cause of chronic diarrhea after solid-organ Am J Transplant. 2011 ;11(3):575-82.
- Skinnider Lymphoproliferative Disorders of the Gastrointestinal Tract. Archives of Pathology & Laboratory Medicine. 2018;142:44-52.
- Van Vliet C, Spagnolo T- and NK-cell lymphoproliferative disorders of the gastrointestinal tract: review and update. Pathology. 2020;52(1):128-41.
- Chott A, Haedicke W, Mosberger I, et al. Most CD56+ intestinal lymphomas are CD8+CD5-T-cell lymphomas of monomorphic small to medium size histology. The American journal of pathology. 1998 ;153(5):1483-90.
- Okuda M, Nomura J, Tateno H, et al. CD56 positive intestinal T-cell lymphoma: treatment with high dose chemotherapy and autologous peripheral blood stem cell Internal medicine (Tokyo, Japan). 2002;41(9):734-7.
- Deleeuw RJ, Zettl A, Klinker E, et al. Whole-genome analysis and HLA genotyping of enteropathy-type T-cell lymphoma reveals 2 distinct lymphoma Gastroenterology. 2007;132 (5):1902-11.
- Carbonnel F, d’Almagne H, Lavergne A, et al. The clinicopathological features of extensive small intestinal CD4 T cell Gut. 1999;45(5):662-7.
- Malamut G, Meresse B, Kaltenbach S, et al. Small Intestinal CD4+ T-cell Lymphoma is a Heterogenous Entity with Common Pathology Clin Gastroenterol Hepatol. 2014;12(4):599-608.
- Koulaouzidis A, Douglas S, Rogers MA, et al. Fecal calprotectin: a selection tool for small bowel capsule endoscopy in suspected IBD with prior negative bi-directional Scand J Gastroenterol. 2011;46(5):561-6.
- Wong G, Lim K, Wan W, et Eosinophilic gastroenteritis: Clinical profiles and treatment outcomes, a retrospective study of 18 adult patients in a Singapore Tertiary Hospital. Med J Malaysia. 2015;70(4):232-7.
- Cunningham-Rundles C, Bodian Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol 1999 ; 92(1) : 34-48.
- Gillon Clinical studies in adults presenting with giardiasis to a gastrointestinal unit. Scott Med J. 1985;30(2):89-95
- Lagier JC, Raoult Whipple’s disease and Tropheryma whipplei infections: when to suspect them and how to diagnose and treat them. Curr Opin Infect Dis. 2018 Dec;31(6):463-470.
- Walker What is tropical sprue? J Gastroenterol Hepatol 2003;18:887-90.
- Rickles FR, Klipstein FA, Tomasini J, et al. Long-term follow-up of antibiotic-treated tropical Annals of internal medicine 1972;76:203-10.
- Gatta L, Scarpignato Systematic review with meta-analysis: Rifaximin is effective and safe for the treatment of small intestine bacterial overgrowth. Aliment Pharmacol Ther. 2017;45:604-16.
- Zhong C, Qu C, Wang B, et al. Probiotics for Preventing and Treating Small Intestinal Bacterial Overgrowth: A Meta-Analysis and Systematic Review of Current J Clin Gastroenterol. 2017;51(4):300-11.
- Guadalupe M, Reay E, Sankaran S, et al. Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration folowing highly active antiretroviral therapy. J Virol. 2003;77(21):11708-17.
- Wang H, Kotler HIV enteropathy and aging: gastrointestinal immunity, mucosal epithelial barrier, and microbial translocation. Curr Opin HIV AIDS. 2014;9(4):309-16.
- Chua I, Standish R, Lear S, et al. Anti-tumour necrosis factor-a therapy for severe enteropathy in patients with common variable immunodeficiency (CVID). Clin Exp Immunol 2007 ; 150(2) : 306-11.
- Parlato M, Charbit-Henrion F, Nader EA, et al. Efficacy of Ruxolitinib Therapy in a Patient with Severe Enterocolitis Associated with a STAT3 Gain of Function Mutation. Gastroenterology 2019;156(4):1206-10
- Forbes LR, Vogel TP, Cooper MA, et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 J Allergy Clin Immunol 2018;142:1665-9.
- Lee S, Moon JS, Lee C-R, et al. Abatacept alleviates severe autoimmune symptoms in a patient carrying a de novo variant in CTLA-4. J Allergy Clin Immunol 2016;137(1):327-30.
- Malamut G, Chandesris C, Verkarre V, et al. Enteropathy Associated T cell Lymphoma in Celiac disease: a large retrospective Dig Liver Dis 2013;45(5):377-84
- Sharma A, Oishi N, Boddicker R, et al. Recurrent STAT3-JAK2 fusions in indolent T-cell lymphoproliferative disorder of the gastrointestinal Blood. 2018;131(20):2262-6.