Liens d’intérêts
Pas de conflit d’intérêt
Mots-clés
- Cancer du pancréas familial
- dépistage
- prédisposition et consultation d’oncogénétique
Abréviations
- Adénocarcinome du pancréas : ADKP
- Cancers pancréatiques familiaux : CaPaFa
Introduction
L’adénocarcinome du pancréas (ADKP) connaît une augmentation d’incidence sans précédent depuis les vingt dernières années et les perspectives publiées sont inquiétantes. En France, de 2006 à 2012, son incidence a doublé passant de 6 000 à plus de 12 000 nouveaux cas par an. Dans une étude récente, les analyses faites estiment un nouveau doublement d’incidence d’ici à 2030 aux États-Unis, mais également en Europe. L’adénocarcinome du pancréas serait la deuxième cause de mortalité par cancer (tous cancers confondus) dès 2020. Les possibilités thérapeutiques de l’ADKP sont très limitées. À ce jour, seule une résection chirurgicale complète et à un stade précoce peut prétendre à une mise en rémission, cela concerne peu de patients car 80 % des ADKP sont diagnostiqués à un stade avancé (1,2).
Pour de nombreux cancers (sein, colorectal, col de l’utérus…), les politiques de dépistage et le traitement des lésions pré-cancéreuses ont permis de diminuer la mortalité. Malheureusement l’ADKP ne rentre pas dans cette catégorie en raison d’une accessibilité limitée à la glande pancréatique, organe profond, et d’un manque d’outils de dépistage fiables, sensibles et non invasifs. Ainsi en population générale, il n’est pas recommandé de dépister le cancer du pancréas. Le dépistage par imagerie est recommandé chez les personnes dites à haut risque d’ADKP (6-3).
Quels sont les facteurs de risque du cancer du pancréas ?
Parmi les facteurs de risque clairement identifiés d’ADKP, on dénombre des facteurs de risque dits environnementaux (tabac, diabète, obésité et pancréatite chronique) et des facteurs constitutionnels, c’est-à-dire des formes à prédisposition familiale (7). On distingue ainsi :
- les formes dites syndromiques, en lien avec une anomalie moléculaire germinale ou constitutionnelle, généralement de transmission autosomique dominante, associée à une augmentation du risque d’autres cancers ;
- les agrégations familiales non syndromiques de cancers du pancréas (également appelés cancers pancréatiques familiaux ou CaPaFa), au sein d’une même famille.
Dépistage et cancer du pancréas, est-ce 2 notions compatibles ?
Dépister suppose :
- un risque de cancer important dans la population cible ;
- de pouvoir mettre en évidence des lésions précancéreuses ou des cancers à un stade précoce accessibles à une chirurgie d’exérèse dans le but de diminuer le risque de décès par cancer ;
- des outils radiologiques ou biologiques (biomarqueurs) fiables, sensibles, reproductibles, accessibles par les centres et non invasifs.
Répondre à ces trois postulats explique qu’il n’y a pas d’indication au dépistage de l’ADKP en population générale :
- L’incidence de l’ADKP ne cesse d’augmenter depuis 20 ans (avec une prévalence proche de l’incidence en raison du fort taux de mortalité) mais cependant l’incidence ne représente « que » 0,5 à 1,3 % sur la durée de vie (4,6).
- Les lésions précancéreuses du pancréas, au nombre de 3, sont les Pancreatic intraepithelial neoplasia (PanIN), les tumeurs intracanalaires papillaires et mucineuses du pancréas (TIPMP) et les cystadénomes mucineux.
- Les cystadénomes mucineux sont des lésions kystiques rares (prévalence inconnue) dont le risque de dégénérescence est inconnu. Cependant le nombre d’AKP développés sur cystadénome mucineux est exceptionnel.
- Les TIPMP sont des lésions kystiques de diagnostic simple en imagerie. Elles sont certes très fréquentes et touchent > 7 % de la population générale adulte. Cependant sachant que seuls 10 % des ADKP se développent sur TIPMP, le risque global de dégénérescence reste très faible. Une résection systématique n’est donc pas recommandée (8).
- Les PanIN sont des lésions microscopiques à partir desquelles se développent 90 % des ADKP mais par définition ne peuvent être diagnostiquées par imagerie (9).
- À ce jour, il n’existe pas encore de biomarqueurs fiables des lésions précancéreuses (PanIN) ou d’ADKP à un stade précoce (i.e : ADN circulant, cellules tumorales circulantes….). Les moyens d’imagerie disponible permettent le dépistage des CM ou des TIPMP mais pas les PanIN de façon fiable.
Le dépistage n’est donc recommandé que pour les individus à haut risque ayant des facteurs de prédisposition au cancer du pancréas (5 à 10 % des cas) (4,6). Ce dépistage n’a pas prouvé son intérêt médico-économique. Les modalités proposées sont encore largement débattues et ont fait l’objet de recommandations internationales par un consortium d’experts [International Cancer of the Pancreas Screening (CAPS) Consortium] publiées
initialement en 2013 (5) et mises à jour en 2020 (6). La même année étaient publiées par 3 experts une mise à jour des recommandations de la société américaine de Gastroentérologie [AGA Clinical Practice Update on Pancreas Cancer Screening in High-Risk Individuals: Expert Review (10)]. Les recommandations sont peu ou prou similaires entre les 2 groupes, les différences de propositions sont à la marge.
Qui sont les patients à haut risque de cancer du pancréas ?
Les individus à haut risque sont les personnes ayant un risque cumulé d’ADKP sur la durée de vie > 5 % ou un risque relatif ≥ 5. Il existe trois catégories de sujets à haut risque :
1. les individus ayant une mutation constitutionnelle dans le cadre d’un syndrome de prédisposition aux cancers.
Les mutations constitutionnelles des gènes de prédisposition actuellement reconnues pour favoriser le cancer du pancréas sont des mutations germinales de la voie BRCA-Fanconi (gènes BRCA1/2, PALB2, ATM, FANC), du gène LKB1/STK11 (s’intégrant dans le syndrome de Peutz- Jeghers), du gène p16/CDKN2A (mélanome familial multiple) ou du syndrome de Lynch (MLH1, MSH2, MSH6, PMS2).
L’ensemble des données est présenté dans le tableau 1 avec le risque relatif de cancer selon le type de mutation.
2. les individus appartenant à une famille de cancer pancréatique familial (CaPaFa) sans mutation identifiée.
Il s’agit de la situation la plus fréquente. En effet il n’y a pas de mutation constitutionnelle trouvée dans 80-85 % des cas où une prédisposition est suspectée. Les individus appartement à cette catégorie ont
- soit ≥ 1 apparenté(s) au 1er degré atteint(s) d’un ADKP ayant lui-même un apparenté au 1er degré atteint d’ADKP (6),
- soit ≥ 3 apparentés atteints d’un ADKP dont un au 1er degré dans la même branche familiale (côté paternel ou maternel) (5,10).
Il est important de noter que le risque de développer un ADKP augmente avec le nombre d’apparentés atteints dans la famille, avec un risque relatif standardisé de 4 à 6 chez les sujets ayant 1 à 2 apparentés atteints, et jusqu’à 20 à 40 chez les sujets ayant ≥ 3 apparentés au 1er degré, atteints de cancer.
3. les individus ayant une pancréatite chronique héréditaire liée à une mutation du gène PRSS1 ((protease serine 1) codant pour le trypsinogène cationique.
Dans l’étude française réalisée à partir de la cohorte nationale de patients porteurs d’une mutation de PRSS1, les risques cumulés de cancer du pancréas à 50, 60 et 75 ans étaient respectivement de 10 % (IC95 %, 1 %-18 %), 18,7 % (IC95 % : 3 %-32 %) et 53,5 % (IC95 % : 7 %-76 %).
La pancréatite chronique héréditaire liée à PRSS1 était associée à un risque élevé relatif et absolu de cancer par rapport à la population générale (SIR : 87 – IC95 % :42-114). Le type de mutation de PRSS1, l’origine de la transmission des mutations de PRSS1 ou la présence d’autres types de mutations des gènes CFTR et SPINK1 n’étaient pas associés à un risque accru de cancer, contrairement au diabète (risque relatif : 13 ; IC95 % :
3-65) et au statut de fumeur. Le risque relatif de cancer chez les fumeurs porteurs d’une mutation de PRSS1 était de 8,55 (IC95 % : 1-70), avec un risque cumulé supérieur à 50 % à 70 ans (11).
Pour les pancréatites chroniques génétiques liées aux autres gènes de prédisposition (SPINK1, CFTR, CTRC, CPA1, CaSR, Cel-HYB, TRPV6), ou pancréatites d’autres causes (alcooliques, autoimmunes…), il n’y a actuellement aucune recommandation de dépistage (6,10,12,13). Cependant des études sont en cours pour les pancréatites génétiques et idiopathiques en raison d’un sur-risque de cancer décrit dans la littérature.
| Situation | Gènes | Fonctionnalité du gène | Spectre tumoral | Pourcentage parmi les ADK avec histoire familiale | Risque ADKP à 70 ans (%) | RR ADKP |
|---|
| Pancréatite Génétique | PRSS1 | Trypsinogène cationique | – | 1-4 | 40-55 | 50-80 |
| Cancer Héréditaire Sein-Ovaire | BRCA2 | Réparation de l’ADN | sein, ovaire, prostate, mélanome | 5-20 | 4-5 | 2-10 |
| BRCA1 | sein, ovaire, colon | 1-5 | 3-4 | 2-4 |
| PALB2 | sein, prostate | 1-3 | 4-5 | 2-6 |
| Mélanome Multiple Héréditaire (FAMMM) | CDKN2A | Régulation du cycle cellulaire | mélanome, sarcome, sein | 2-3 | 5-25 | 10-25 |
| Syndrome de Peutz-Jeghers | LKB1/ STK11 | Régulation de l’apoptose, voie du VEGF | sein, colorectal, estomac | 1-3 | 30-40 | 100-130 |
| Syndrome de Lynch | MLH1, MSH2, MSH6, PMS2 | Réparation de l’ADN | colorectal, utérus, voies urinaires, grêle, cholangiocarcinome, cerveau | 1-3 | 3-5 | 4-8 |
| Polypose Adénomateuse Familiale | APC | Adhérence cellulaire | colorectal, estomac, tumeurs desmoïdes | <2 | ? | ? |
| Ataxie-Télangiectasie | ATM mono- allélique | Réparation de l’ADN | sein, prostate, estomac, hémopathie | <2 | ? | ? |
| Syndrome de Li-Fraumeni | TP53 | Réparation de l’ADN, contrôle du cycle cellulaire et de l’apoptose | sarcome, sein, cerveau, hémopathie | <2 | ? | ? |
| 2 apparentés au 1er degré atteint d’ADKP | ? | – | – | 80-85 | 5-12 | 4-6 |
| 3 apparentés au 1er degré atteint d’ADKP | ? | – | – | 40 | 20-40 |
| Pas d’antécédent familial | – | – | – | – | 0,5-1 | 1 |
La consultation d’Oncogénétique
En cas de suspicion de forme syndromique ou de cancer pancréatique familial, une consultation d’oncogénétique doit être proposée au patient. L’objectif d’une consultation d’oncogénétique est de poser le diagnostic de cancer de forme héréditaire et/ou de faire une étude moléculaire afin d’identifier la mutation causale, et ainsi :
- confirmer, sur une base moléculaire, le diagnostic évoqué,
- proposer un protocole de dépistage adapté,
- proposer la réalisation d’un test moléculaire ciblé aux apparentés à risque (si l’anomalie moléculaire a été mise en évidence).
À noter que l’absence d’identification d’une anomalie constitutionnelle ne permet pas d’éliminer le diagnostic de forme héréditaire. Ce diagnostic est alors basé sur des critères généalogiques, les antécédents personnels et familiaux du patient. Le déterminisme n’est alors pas connu (altération d’un ou de plusieurs gènes non encore identifiés).
Au cours de cette consultation dédiée faite par un onco-généticien, il est réalisé un arbre généalogique détaillé. Un prélèvement sanguin est effectué pour recherche de mutations constitutionnelles sous forme d’un panel de gènes. En cas de mutation délétère mise en évidence, un second prélèvement est nécessaire pour confirmer le résultat (prélèvement jugal ou sanguin). La découverte d’une mutation constitutionnelle impacte le patient mais aussi tous ses apparentés et conduit à une prise en charge multidisciplinaire afin de réaliser si nécessaire un dépistage d’autres cancers associés (sein, ovaire, colon, mélanome, etc.). Le patient doit recevoir une information claire et détaillée avant de réaliser le prélèvement. Son consentement écrit est requis.
Au final, à qui et quand proposer un dépistage du cancer du pancréas ? Les dernières recommandations d’experts du CAPS consortium (6) et de l’AGA (10)
Un programme de dépistage peut être proposé à :
- Tout patient atteint d’un syndrome de Peutz-Jeghers, c’est-à-dire porteur d’une mutation germinale du gène LKB1/STK11, indépendamment des antécédents familiaux.
- Tout patient porteur d’une mutation germinale de CDKN2A, indépendamment des antécédents familiaux.
- Tout patient ayant une pancréatite chronique héréditaire secondaire à une mutation de PRSS1, indépendamment des antécédents familiaux.
- Tout porteur d’une mutation germinale des gènes BRCA2, BRCA1, PALB2, ATM, MLH1, MSH2 ou MSH6 avec au minimum 1 parent au 1er degré atteint d’ADKP.
Remarque : dans les 2 articles (6,10), il n’est pas clairement mentionné si les porteurs de mutations des gènes TP53 et APC sont intégrés dans cette catégorie. En pratique, ils le sont en cas de mutation présente associée à un antécédent familial au 1e degré d’ADKP. - Les personnes ayant au moins un parent au 1er degré atteint d’un ADKP ayant lui-même un parent au 1er degré atteint d’ADKP.
- Les personnes ayant 3 antécédents familiaux d’ADKP dont un au 1er degré.
Tous les individus répondant aux critères ci-dessus sont éligibles à un dépistage. Il faut cependant garder en mémoire ses limites, trouver le juste équilibre et ne proposer ce programme qu’aux patients candidats à une éventuelle chirurgie pancréatique en tenant compte de leur espérance de vie théorique et co-morbidités (6,10).
Quand débuter le dépistage ?
- Pour les cancers pancréatiques familiaux (agrégations de cas) : Le dépistage est proposé à partir de 50 ans ou 10 ans avant le cas index le plus jeune de la famille.
- Pour les individus atteints de pancréatite chronique héréditaire (PRSS1), de syndrome de Peutz-Jeghers (LKB1/STK11) ou de mélanomes multiples (CDKN2A), le dépistage est recommandé dès l’âge de 40 ans (5,6,10).
- Pour les porteurs d’une mutation germinale des gènes BRCA2, BRCA1, PALB2, ATM, MLH1, MSH2 ou MSH6 avec au moins 1 parent au premier degré atteint d’ADKP : dépistage dès l’âge de 45 ans ou 10 ans avant le cas index familial.
Il n’y a pas de consensus sur l’âge de fin de la surveillance. Cependant le dépistage doit être arrêté si le patient n’est plus éligible à un geste chirurgical éventuel ou si le risque de décès d’une cause extra-pancréatique est supérieur au risque de décès par ADKP (6,10).
Quel dépistage proposer pour les patients à haut risque ?
Actuellement, le dépistage repose sur les examens d’imagerie : échoendoscopie et IRM pancréatiques. Le but est de chercher des lésions pancréatiques solides ou kystiques. Le TDM est privilégié uniquement pour les patients pour lesquels l’IRM et EUS sont contre-indiquées. L’IRM est préférée au TDM en raison de sa plus grande sensibilité pour détecter les lésions kystiques de petite taille, millimétriques (14) et pour limiter l’irradiation des patients. Même si les kystes pancréatiques (microkystes non branchés et TIPMP des canaux secondaires) ont un faible potentiel de malignité, leur détection permet une stratification du risque (8). L’IRM permet également la mise en évidence de lésion tumorale solide, principal but de ce dépistage.
L’EUS est aussi un examen clé pour détecter les petites lésions d’ADKP. L’expertise du praticien est majeure et ce dépistage doit être confié à des opérateurs expérimentés. L’EUS permet d’identifier également des anomalies parenchymateuses indirectement liées à la présence de PanIN, tels que des spots d’atrophie focale et des défauts de lobularité (9).
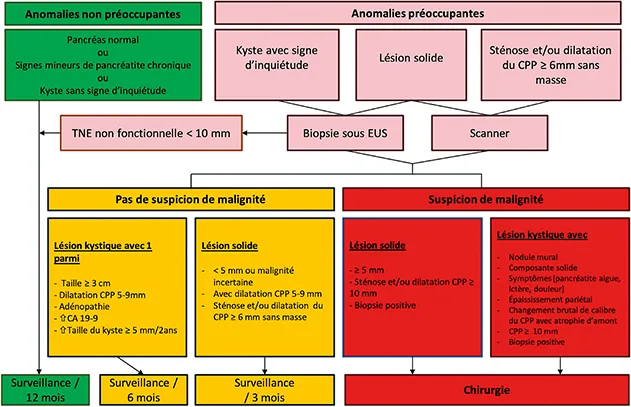
Les experts ne sont pas parvenus à un consensus quant aux modalités sur le fait d’alterner ou non ces deux examens au cours du suivi. Les experts préconisent le schéma de dépistage suivant… (page ci-contre).
Points à souligner
- la fréquence des examens est à moduler en fonction des anomalies mises en évidence,
- en l’absence d’anomalies, la fréquence des examens est annuelle,
- le TDM a un intérêt en cas de lésion solide ou de sténose du canal pancréatique principal pour compléter le bilan lésionnel avant de décider d’un geste de résection,
- le Ca 19-9 n’est pas recommandé dans le suivi et n’est pas à doser,
- la recherche d’un trouble de la glycorégulation (dosage annuel de la glycémie à jeun ou de l’hémoglobine glyquée) est recommandé. En effet, plusieurs études ont clairement montré que le diabète pouvait être un signe précurseur d’apparition de l’ADKP, notamment dans les 3 ans précédant le diagnostic de cancer (15). Il ne s’agit pas alors d’un diabète de type 1, 2 ou 3 mais d’un diabète considéré comme « paranéoplasique ». Le mécanisme physiopathologique sous-jacent n’est absolument pas compris à ce jour. De même toute décompensation diabétique (d’un diabète connu ancien de type 1 ou 2), par analogie, est un point d’appel qui doit motiver une nouvelle imagerie en urgence et ainsi devancer les examens prévus dans l’année.

Schéma de dépistage préconisé par les experts
Conclusion
Beaucoup d’inconnues persistent quant au dépistage du cancer du pancréas dans les populations dites à haut risque. Les schémas de dépistage publiés ne sont que des recommandations d’experts car les études de haut niveau de preuve n’existent pas. Ainsi ce dépistage reste débattu, aucune étude n’a prouvé son intérêt médico-économique. Il est primordial
- de réaliser ce dépistage dans un centre maîtrisant les modalités et les attendus de l’IRM et l’échoendoscopie pancréatiques dans ce contexte,
- de discuter de ces situations complexes dans le cadre de réunion de concertation pluridisciplinaire,
- d’expliquer au patient les limites de ce dépistage,
- de prendre en considération le caractère anxiogène de ces programmes et de proposer un soutien psychologique au patient si besoin,
- d’informer le patient et prendre en charge tous les facteurs de risque de cancer associés qui pourraient augmenter le risque : prévention du tabac, de l’obésité et de la sédentarité.
Références
- Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74(11):2913-21.
- Neuzillet C, Tijeras-Raballand A, Bourget P, et al. State of the art and future directions of pancreatic ductal adenocarcinoma therapy. Pharmacol Ther 2015;155:80-104.
- Aslanian HR, Lee JH, Canto MI. AGA Clinical Practice Update on Pancreas Cancer Screening in High-Risk Individuals: Expert Review. Gastroenterology 2020 Jul;159(1):358-362.
- Neuzillet C, Gaujoux S, Williet N, et al. Pancreatic cancer: French clinical practice guidelines for diagnosis, treatment and follow-up (SNFGE, FFCD, GERCOR, UNICANCER, SFCD, SFED, SFRO, ACHBT, AFC). Dig Liver Dis Off J Ital Soc Gastroenterol Ital Assoc Study Liver 2018;50(12):1257-71.
- Canto MI, Harinck F, Hruban RH, et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013;62(3):339-47.
- Goggins M, Overbeek KA, Brand R, et al. Management of patients with increased risk for familial pancreatic cancer: updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut 2020;69(1):7-17.
- Barone E, Corrado A, Gemignani F, Landi S. Environmental risk factors for pancreatic cancer: an update. Arch Toxicol. 2016 Nov;90(11):2617- 2642.
- European Study Group on Cystic Tumours of the Pancreas. European evidence-based guidelines on pancreatic cystic neoplasms. Gut 2018;67(5):789-804.
- Brune K, Abe T, Canto M, et al. Multifocal neoplastic precursor lesions associated with lobular atrophy of the pancreas in patients having a strong family history of pancreatic cancer. Am J Surg Pathol 2006;30(9):1067-76.
- Aslanian H.R, Lee J.H, Canto I.C. AGA Clinical Practice Update on Pancreas Cancer Screening in High-Risk Individuals: Expert Review. Gastroenterology. 2020 Jul;159(1):358-362.
- Rebours V, Boutron-Ruault MC, Schnee, et al. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: a national exhaustive series. Am J Gastroenterol. 2008 Jan;103(1):111-9.
- Muller N, Sarantitis I, Rouanet M, et al. Natural history of SPINK1 germline mutation related-pancreatitis.EBioMedicine. 2019 Oct;48:581- 591.
- Greenhalf W, Lévy P, Gress T, et al. Consensus Guidelines for Chronic Pancreatitis.International consensus guidelines on surveillance for pancreatic cancer in chronic pancreatitis. Recommendations from the working group for the international consensus guidelines for chronic pancreatitis in collaboration with the International Association of Pancreatology, the American Pancreatic Association, the Japan Pancreas Society, and European Pancreatic Club. Pancreatology. 2020 Jul;20(5):910-918.
- Vullierme M-P, Menassa L, Couvelard A, et al. Non-branched microcysts of the pancreas on MR imaging of patients with pancreatic tumors who had pancreatectomy may predict the presence of pancreatic intraepithelial neoplasia (PanIN): a preliminary study. Eur Radiol 2019.
- Huang B, Pandol S, Jeon C, et al. New-Onset Diabetes, Longitudinal Trends in Metabolic Markers, and Risk of Pancreatic Cancer in a Heterogeneous Population. Clinical Gastroenterology and Hepatology 2020;18:1812–1821