Liens d’intérêt
L’auteure n’a pas déclaré de conflit d’intérêt en lien avec sa présentation
Mots-clés
Hypertension portale ; anticoagulants ; maladie rare
Abréviations
MVPS : maladie vasculaire porto-sinusoïdale
SOS : syndrome d’obstruction sinusoîdale
SBC : syndrome de Budd-Chiari
TVP : thrombose de la veine porte
SMP : syndrome myéloprolifératif
TIPS : shunt portocave intra-hépatique transjugulaire
HTP : hypertension portale
HBPM : héparine de bas poids moléculaires
AVK : antivitamines K
AOD : anticoagulants oraux directs
ETP : Education thérapeutique
Introduction
Les maladies vasculaires du foie regroupent un large éventail d’entités clinico-pathologiques résultant d’une atteinte du système vasculaire hépatique. Elles comprennent les thromboses veineuses splanchniques (syndrome de Budd-Chiari et thrombose de la veine porte), les atteintes des petits vaisseaux et les maladies de l’artère hépatique. Il s’agit dans la majorité des cas de maladies rares, définies par une prévalence inférieure à ½ 000 habitants (1). La principale manifestation des maladies vasculaires du foie est l’hypertension portale. Dans ce chapitre, nous détaillerons les manifestations et la prise en charge du syndrome de Budd-Chiari et de la maladie vasculaire porto-sinusoïdale. Le diagnostic et la prise en charge de la thrombose de la veine porte ne seront pas traités.
Classification des maladies vasculaires du foie
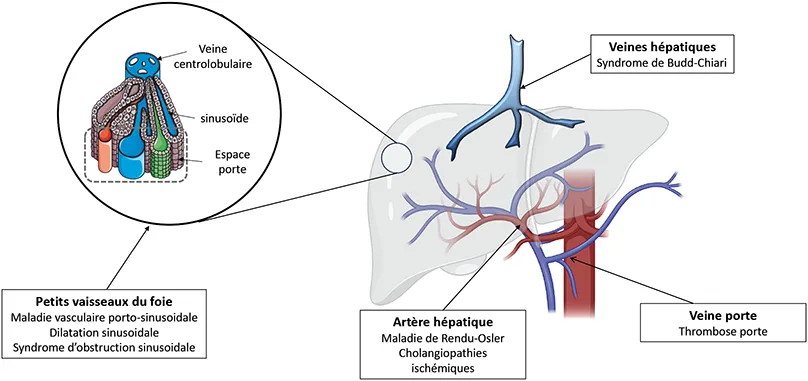
Les maladies vasculaires du foie peuvent être classées en plusieurs catégories : (i) les atteintes veineuses macroscopiques, (ii) les atteintes microvasculaires, (iii) les atteintes artérielles, (iv) les fistules (2) (Figure 1).
Les obstructions veineuses macroscopiques comprennent la thrombose de la veine porte (TVP) en l’absence de cirrhose (non traité ici) et le syndrome de Budd-Chiari (obstacle au retour veineux hépatique, le plus souvent lié à une thrombose des veines hépatiques ou de la veine cave inférieure). Ce sont des maladies rares, qui partagent de nombreux facteurs de risque parmi lesquels le syndrome myéloprolifératif est le plus fréquent.

Figure 1 : Classification des maladies vasculaires du foie
Les maladies des petits vaisseaux du foie regroupent des lésions des petits vaisseaux du foie, au niveau de l’espace porte, du sinusoïde hépatique ou de la veine centro-lobulaire. Les atteintes microvasculaires hépatiques comprennent la maladie vasculaire porto-sinusoïdale (MVPS), la distension sinusoïdale et le syndrome d’obstruction sinusoïdale (SOS). D’un point de vue histologique, les lésions de la MVPS sont primitivement situées au niveau de l’espace porte alors que les lésions du SOS sont primitivement sinusoïdales et centrolobulaires.
Les atteintes artérielles hépatiques comprennent la maladie de Rendu-Osler et les cholangiopathie ischémiques (lésions focales ou étendues des canaux biliaires liées à un trouble de leur vascularisation artérielle).
Enfin, la fistule portosystémique congénitale (FPC), également appelée « malformation d’Abernethy », est une malformation congénitale définie par une ou plusieurs communications entre une veine du système veineux portal à destinée hépatique et une veine du système veineux cave à destinée systémique. Dans la grande majorité des cas, le diagnostic est posé dans la petite enfance ou dans l’enfance. Les principales manifestations sont liées aux conséquences des shunts : encéphalopathie hépatique, les complications cardiaques et/ou pulmonaires, et tumeurs hépatiques bénignes ou malignes (3).
Syndrome de Budd Chiari
Épidémiologie et causes
Le syndrome de Budd-Chiari (SBC) est causé par un obstacle au retour veineux hépatique qui peut se situer depuis les petites veines hépatiques jusqu’à l’abouchement de la veine cave inférieure dans l’oreillette droite. Le SBC est généralement causé par une thrombose. C’est une maladie très rare (incidence d’un cas par million d’habitants par an), affectant principalement des sujets d’âge jeune (âge médian au diagnostic de 35-40 ans), avec une prédominance féminine (4).
Le SBC est la plupart du temps primitif, l’obstruction étant liée à une thrombose ou sténose. Le SBC secondaire, causé par une compression extrinsèque par une tumeur, un abcès ou un kyste, est plus rare.
Un bilan étiologique approfondi à la recherche d’une maladie pro-thrombotique doit être systématiquement réalisé (tableau 1).
La cause la plus fréquente de SBC est le syndrome myéloprolifératif (SMP) (plus de 40 % des cas). La mutation JAK2 V617F doit être systématiquement cherchée. Elle est présente chez 80-90 % des malades atteints de SBC et de SMP. En l’absence de mutation JAK2 V617F, d’autres mutations doivent être recherchées : une mutation somatique du gène codant pour la calréticuline (CALR) – surtout si le taux de plaquettes est > 200 x 10^9/L avec une splénomégalie > 16 cm (5) –, une mutation de l’exon 12 de JAK2, et une mutation du récepteur à la thrombopoïétine (MPL) (4). Si ces mutations sont absentes, un séquençage à haut débit [également appelé next generation sequencing (NGS)] et une biopsie ostéomédullaire peuvent être discutés en concertation avec les hématologues pour diagnostiquer un potentiel syndrome myéloprolifératif sous-jacent (6).
Une thrombophilie héréditaire doit également être recherchée, la mutation G20210A du gène de la prothrombine est la plus fréquente (7).
Les autres causes de SBC sont des états prothrombotiques acquis : le syndrome des anticorps antiphospholipides (SAPL), l’hémoglobinurie paroxystique nocturne, la maladie de Behçet, la maladie cœliaque et des facteurs hormonaux (grossesse, contraception orale œstro-progestatives) (4, 7). La combinaison d’au moins deux facteurs de risque génétiques ou acquis est présente chez 26 % à 46 % des patients, ce qui justifie un bilan exhaustif, même lorsqu’un facteur favorisant a déjà été mis en évidence (8).
Tableau 1 : Prévalence des facteurs de risque du syndrome de Budd-Chiari (SBC) en l’absence de maladie du foie sous-jacente et bilan diagnostic recommandé [d’après (7)]
| Condition | Prévalence | Bilan recommandé en 1ère intention |
| Syndrome myéloprolifératif |
| JAK2 V617F | 28-45 % | Test génétique de la mutation V617F du gène JAK2 chez tous les patients. Si négatif – test génétique du gène de la calréticuline si le nombre de plaquettes est ≥ 200 x109 /L et/ou la hauteur de la rate ≥ 16 cm. |
| Mutation CALR | 1-3 % |
| Thrombophilies héréditaires |
| Mutation du gène de la prothrombine G20210A | 12 % | Tests génétiques – Mutation du gène de la prothrombine G20210A – Mutation du facteur V Leiden Activité de l’antithrombine Activité de la protéine C Activité de la protéine S Le dosage doit être effectué en l’absence d’AVK. L’interprétation doit être prudente en cas d’altération de la fonction hépatique |
| Mutation du facteur V Leiden | 4 % |
| Déficit en antithrombine | 3 % |
| Déficit en protéine C | 2 % |
| Déficit en protéine S | 2 % |
| États prothrombotiques acquis |
| Syndrome des anticorps antiphospholipides | 5 % | Recherche d’anticorps anticoagulant circulant lupique, anti-cardiolipine et anti-bêta2 glycoprotéine 1 Répétition du test après 12 semaines en cas de test positif |
| Hémoglobinurie paroxystique nocturne | 10 % | Analyse par cytométrie en flux |
| Maladie de Behçet | 1-2 % | Pas de test spécifique, diagnostic clinique Éléments évocateurs : sexe masculin, origine méditerranéenne, sténose de la veine cave inférieure, ulcères génitaux/buccaux, thrombose veineuse profonde dans d’autres sites, thrombose artérielle |
| Maladie cœliaque | 1-4 % | Anticorps anti-transglutaminase +/- biopsies duodénales |
| Autres |
| Facteurs hormonaux Contraceptif oral ou grossesse | ≈ 30 % | Contexte clinique Introduction/modification dans les 6 mois précédant le diagnostic |
| Aucun facteur identifié | 10-30 % | |
AVK : antagonistes de la vitamine K ; CALR : calréticuline ; JAK2 : janus kinase 2
Diagnostic du SBC
La présentation clinique du SBC est extrêmement variable, allant de patients asymptomatiques (3 % des cas) à des patients ayant une hypertension portale sévère ou une insuffisance hépatique, voire même une hépatite fulminante. Chez la plupart des patients, les manifestations cliniques comprennent des douleurs abdominales et une ascite (8). La difficulté du diagnostic du SBC est illustrée par une étude épidémiologique française où la durée entre les premiers symptômes et le diagnostic de SBC primitif dépassait 6 mois chez 15 % des patients et un an chez 6 % des patients (3). En pratique, un SBC doit être évoqué chez tout patient atteint de maladie hépatique aiguë ou chronique (9).
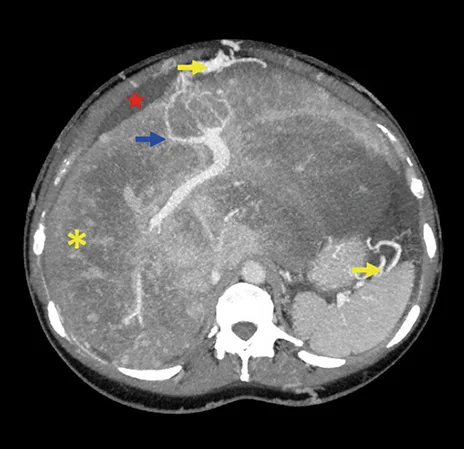
Le diagnostic de SBC repose sur l’imagerie (échographie abdominale couplée à un doppler, complétée par une TDM ou IRM hépatique avec injection de produit de contraste). Les signes radiologiques de SBC comprennent (i) des arguments directs d’obstruction, notamment la mise en évidence de matériel endoluminal solide non rehaussé ou la transformation des veines en un cordon fibreux sans flux, et (ii) des signes indirects d’obstruction à l’écoulement du sang dans les veines hépatiques notamment une hépatomégalie congestive, des collatérales veineuses inter- hépatiques, une atrophie des segments atteints et une hypertrophie des segments non atteints, des signes d’hypertension portale (2, 4) (Figure 2). Les formes « classiques » de SBC ne nécessitent pas de biopsie hépatique pour le diagnostic, alors qu’une biopsie peut être utile dans les rares cas de SBC des petites veines hépatiques où les veines hépatiques sont macroscopiquement perméables.
Prise en charge
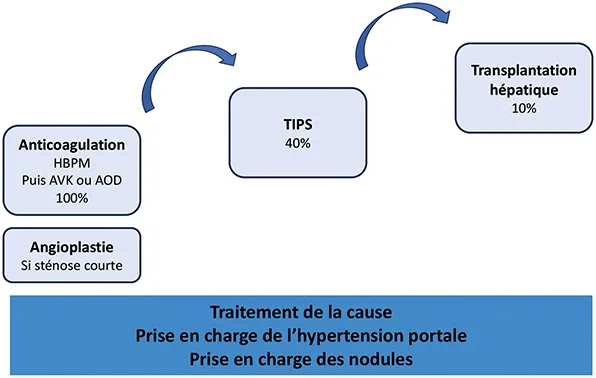
La prise en charge des malades atteints de SBC doit être discutée en concertation avec un centre expert des maladies vasculaires du foie (centre de compétence et/ou centre de référence) ayant accès à la radiologie interventionnelle et à la transplantation hépatique. L’approche doit être multidisciplinaire et inclure des spécialistes de l’hémostase, de l’hématologie, de la radiologie diagnostique et interventionnelle et de la transplantation hépatique. Depuis plus de 15 ans, une stratégie de traitement pas à pas, en fonction de la réponse au traitement précédent (du moins au plus invasif), a été proposée et est maintenant largement utilisée dans le monde entier (10, 11) (figure 3). Grâce à cette stratégie, le pronostic des patients s’est considérablement amélioré, avec une survie globale à 5 ans supérieure à 80 % (11).

Figure 2 : Aspect à la tomodensitométrie d’un patient atteint de syndrome de Budd-Chiari.
Scanner hépatique au temps portal : les veines hépatiques ne sont pas visualisées. Il existe une hépatomégalie congestive (astérisque jaune), et des collatérales inter sus-hépatiques (flèche bleue), ainsi que des collatérale porto-systémiques (flèche jaune) et de l’ascite (étoile rouge), témoignant d’une hypertension portale.
Traitement médical
Il est essentiel d’identifier rapidement les maladies associées (SMP, hémoglobinurie paroxystique nocturne, maladie de Behçet), car l’introduction d’un traitement spécifique de la maladie associée peut faire évoluer favorablement certains patients (12, 13).
L’anticoagulation doit être mise en place dès le diagnostic du SBC pour tous les patients, même en l’absence d’état pro-thrombotique identifié. En raison d’un taux élevé de thrombocytopénie induite par l’héparine, principalement observé avec l’héparine non fractionnée (15 %), le traitement par héparine de bas poids moléculaire (HBPM) est recommandé en première intention au diagnostic (14). L’HBPM est généralement remplacée par des AVK chez les patients dont la maladie est stable, avec pour objectif un INR entre 2 et 3. Bien que l’expérience soit limitée, les anticoagulants oraux directs (AOD) semblent sûrs et efficaces chez les patients atteints de SBC (15). Une anticoagulation au long cours est indiquée pour tous les patients atteints de SBC. Cependant, la pharmacocinétique des AOD chez les patients porteurs d’un TIPS n’est pas connue ; par conséquent les AOD ne peuvent être recommandés dans cette situation. Enfin, les AOD doivent être utilisés avec prudence ou même contre-indiqués en cas d’insuffisance hépatique sévère (classe de Child-Pugh B et C) ou d’insuffisance rénale (16).
La prise en charge des complications de l’hypertension portale (ascite, varices gastro-œsophagiennes), de l’insuffisance rénale et de l’encéphalopathie hépatique est similaire à celle recommandée chez les patients atteints de cirrhose (17).

Figure 3 : Algorithme de prise en charge du Syndrome de Budd-Chiari
HBPM : héparine de bas poids moléculaire ; AVK : antivitamines K ; AOD : anticoagulants oraux directs ; TIPS : shunt intrahépatique porto-systémique transjugulaire.
Radiologie interventionnelle
Lorsqu’une sténose courte d’une veine hépatique est identifiée (environ 15 % des patients), une angioplastie transluminale percutanée de la sténose doit être systématiquement réalisée, car cette procédure a une bonne efficacité et une faible morbidité(18).
Le shunt intrahépatique porto-systémique transjugulaire (TIPS) est proposé chez les patients ayant une réponse incomplète au traitement médical et/ ou à l’angioplastie (environ 40 % des patients) (11). Le taux de réussite technique dépasse 90 % dans les centres experts et la survie sans transplantation hépatique à 10 ans atteint 78 % (11). À l’instar de la cirrhose, le TIPS n’est pas contre-indiqué en cas d’insuffisance hépatique sévère. Même, à l’inverse, une insuffisance hépatique sévère doit faire envisager un TIPS, car celui-ci est associé à une amélioration de la fonction hépatique (11, 19, 20).
Transplantation hépatique
Malgré le traitement médical et la radiologie interventionnelle, la transplantation hépatique est nécessaire pour environ 10 % des patients atteints de SBC, avec une survie à 5 ans de 71 % (11). La présence d’un TIPS ne compromet pas l’accès à la transplantation, ni les résultats de la transplantation hépatique (21).
Diagnostic et prise en charge des nodules
Les nodules hépatiques sont fréquents au cours du SBC (environ 60 % des patients). La plupart sont bénins mais des adénomes hépatocellulaires ou des carcinomes hépatocellulaires (CHC) peuvent également apparaître. L’incidence cumulée du CHC est d’environ 4 %, similaire à celle rapportée dans d’autres maladies chroniques du foie (22). Un dépistage des nodules hépatiques par imagerie tous les 6 mois est donc recommandée (2, 17).
Devant la présence d’un nodule hépatique, l’IRM, idéalement avec injection d’un produit de contraste hépato-spécifique, est l’imagerie de choix pour distinguer les nodules malins des nodules bénins. La grande majorité des nodules présentent une hyper-intensité à la phase artérielle. Le lavage au temps portal (la tumeur apparaît alors hypodense par rapport au parenchyme hépatique avoisinant du fait de la perte de vascularisation portale) est observé dans 75 % des CHC contre 29 % des lésions bénignes et n’est donc pas spécifique du CHC, comme c’est le cas dans la cirrhose. D’autres caractéristiques aident à différencier les CHC des nodules bénins, notamment le contenu graisseux du nodule, la présence d’une capsule, une hypo-intensité sur la séquence pondérée en T1, une hyper-intensité sur la séquence pondérée en T2 ou une hypo-intensité sur la phase hépatobiliaire
(23) et biologiquement, l’élévation de la concentration sérique d’alpha-fœtoprotéine sérique > 15 ng/mL. Une biopsie du foie est nécessaire en cas de suspicion de CHC. La prise en charge des patients ayant un adénome hépatocellulaire ou un CHC doit être discutée au cas par cas dans des centres experts des maladies vasculaires du foie. L’ablation percutanée, la chimioembolisation et la transplantation hépatique peuvent être envisagées. Le TIPS est associé à une morbidité accrue après chimioembolisation artérielle hépatique.
Maladie vasculaire porto-sinusoïdale
Définition
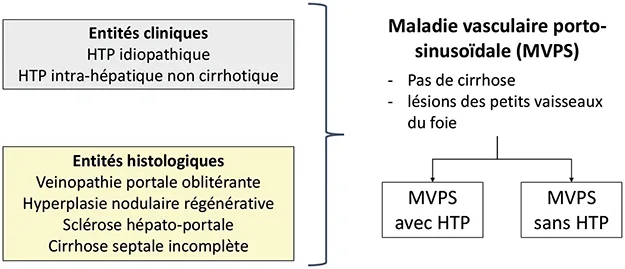
La MVPS regroupe diverses maladies caractérisées par des anomalies des petits vaisseaux du foie. Les terminologies précédemment utilisées figurent dans la figure 4. Il existe un chevauchement entre toutes ces entités et aucune conséquence pratique à les discriminer.
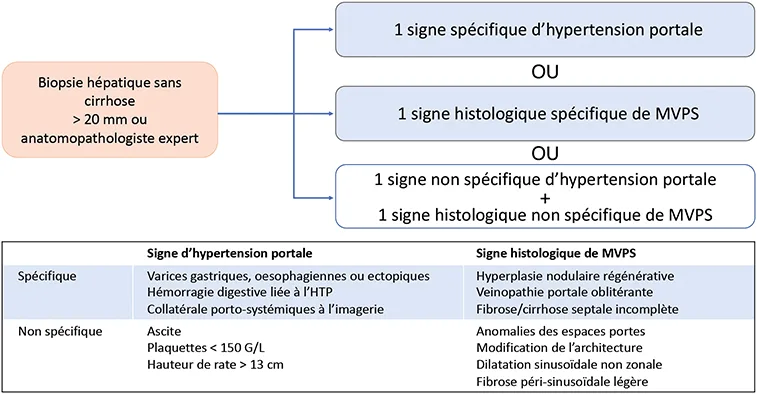
La biopsie hépatique est indispensable pour le diagnostic de MVPS. Ce diagnostic repose, selon le réseau Européen des maladies vasculaires du foie VALDIG, sur (i) l’absence de cirrhose et (ii) des lésions histologiques spécifiques ou des signes d’hypertension portale (Figure 5) (17, 24). La biopsie hépatique est considérée comme adéquate si elle mesure plus de 20 mm de long et contient plus de 10 espaces portes, est peu ou pas fragmentée ou si jugée comme telle par un anatomopathologiste expert. Le diagnostic de MVPS est considéré comme un diagnostic difficile ; plusieurs biopsies sont souvent nécessaires pour poser le diagnostic (25). Il existe deux formes de MVPS : MVPS avec hypertension portale, précédemment nommée « hypertension portale idiopathique », ou « hypertension portale intrahépatique non cirrhotique », et MVPS sans hypertension portale.

Figure 4 : Entités regroupées sous le terme de maladie vasculaire porto-sinusoïdale.
HTP : hypertension portale

Figure 5 : Critères diagnostiques de MVPS [adapté de (26)]
Quand suspecter une MVPS ?
Les deux principaux modes de découverte de la MVPS sont des anomalies chroniques et inexpliquées des tests hépatiques et une hypertension portale sans cirrhose. Dans les deux cas, l’attention doit être attirée par l’absence de cause reconnue d’hépatopathie chronique, telle qu’une consommation excessive de boissons alcoolisées, un syndrome métabolique, une infection par le virus de l’hépatite B ou de l’hépatite C, une surcharge en fer, une hépatite auto-immune ou une obstruction des veines hépatiques. Cependant, la présence d’une cause d’hépatopathie chronique n’exclut pas le diagnostic de MVPS (26).
Les altérations des tests hépatiques associées à la MVPS sont variées. Il s’agit chez d’élévation des transaminases le plus souvent modérée, d’élévation des phosphatases alcalines, parfois supérieure à 2 fois la limite supérieure de la normale, et d’élévation des 0-GT (27, 28).
L’autre manifestation de la MVPS est l’hypertension portale. En cas d’hypertension portale, certains signes doivent faire suspecter une MVPS et encourager à la réalisation d’une ponction-biopsie hépatique. Un contraste entre une hypertension portale franche et des tests de fonction hépatique normaux ou quasi-normaux sont évocateurs de MVPS. Le taux de prothrombine est généralement supérieur à 50 % chez les malades avec MVPS. L’autre tableau qui doit alerter est le contraste entre les signes d’hypertension portale et une élasticité hépatique basse (Fibroscan® < 10 kPa). En effet, dans une étude récente portant sur 155 malades atteints de MVPS et 247 malades atteints de cirrhose compensée, la valeur prédictive positive d’une élasticité hépatique < 10 kPa pour le diagnostic de MVPS était de 92 % (29). Enfin, si le scanner hépatique montre un foie de contours lisses et l’absence d’atrophie du segment IV, il faut suspecter une MVPS. En effet, la combinaison de ces deux caractéristiques avait une VPP de 73 % pour le diagnostic de MVPS dans une étude comportant 198 malades atteints de MVPS et 426 atteints de cirrhose (30).
Atteintes extra-hépatiques associées à la maladie vasculaire porto-sinusoïdale
De nombreuses affections ont été rapportées associées à l’existence d’une MVPS. Elles sont résumées dans le tableau 2. Malgré un bilan exhaustif, aucune condition n’est retrouvée dans 30 % des cas environ. Les examens reposent sur le contexte clinique.
Tableau 2 : Affections extra-hépatiques associées à la MVPS
| Infection |
| – VIH – Angiocholites à répétition |
| Maladies dysimmunitaires ou de système |
| – Déficit immun commun variable – Transplantation d’organe solide (rein, foie) – Maladie de Basedow – Polyarthrite rhumatoïde – Syndrome POEMS – Lupus érythémateux disséminé – Maladie de Wegener – Syndrome de Sharp |
| Maladie hématologique |
| – Myélome multiple – Maladie de Waldenström – Syndrome myélodysplasique – Syndrome myéloprolifératif – Maladie de Hodgkin – Lymphome B marginal – Purpura idiopathique thrombopénique |
| États prothrombotiques |
| – Mutation du gène du facteur II – Mutation du gène du facteur V – Syndrome des anticorps antiphospholipides – Déficit en protéine S – Déficit en protéine C |
| Médicaments (tous débatus) |
| – Didanosine – Azathioprine, – 6-thioguanine, – Arsenic |
| Maladies génétiques |
| – Syndrome d’Adams-Oliver – Syndrome de Turner – Mutation des gènes des télomérases (TERT/TERC) – Formes familiales |
Prise en charge
MVPS sans hypertension portale
En l’absence d’hypertension portale, le risque de survenue de complications (décompensation ou thrombose de la veine porte) est extrêmement faible (31). Cependant, certains malades peuvent développer une hypertension portale et/ou une thrombose porte ; pour cette raison, un suivi reste nécessaire (25). Les modalités de suivi des malades atteints de MVPS sans hypertension portale ne sont pas clairement définies. Une surveillance clinique et biologique, voire par endoscopie ou échographie abdominale peut être proposée, dans le but de dépister la survenue de signes d’hypertension portale qui modifieraient alors le suivi et la prise en charge.
MVPS avec hypertension portale
Traitement médical
L’hémorragie digestive par rupture de varices est l’évènement clinique le plus fréquent, touchant près de la moitié des malades qui ne reçoivent pas de prophylaxie. Même lorsqu’une prophylaxie de la rupture de varices œsophagiennes est mise en place, le risque d’hémorragie digestive est de 25 % à 5 ans. La mortalité causée par une hémorragie digestive par rupture de varices est faible, 3 % à 6 semaines, probablement compte tenu de l’absence d’insuffisance hépato-cellulaire (32).
L’ascite survient le plus souvent au cours d’évènements intercurrents tels qu’une infection ou une hémorragie digestive et est associée à un risque accru de décès (33). Certains malades peuvent développer une ascite réfractaire, surtout en cas d’insuffisance rénale associée (34).
L’encéphalopathie hépatique est une manifestation encore plus rare. En l’absence d’étude concernant spécifiquement cette maladie, il est recommandé de traiter les varices gastro-œsophagiennes selon les mêmes modalités que chez les malades avec cirrhose (17). Comme l’élasticité hépatique est basse, cette mesure ne peut être utilisée pour le dépistage des varices gastro-œsophagiennes (29). En revanche, des données récentes suggèrent que l’association d’une dureté splénique par Fibroscan associée à un taux de bilirubine plasmatique basse (< 17 µmol/L) permet d’écarter des varices à risque de saignement (35).
Après l’hémorragie digestive, la TVP est l’évènement le plus fréquent, avec une incidence de 30 % à 5 ans. Les malades ayant une MVPS associée à une infection VIH ont un risque plus élevé de TVP (32). Une surveillance radiologique de la perméabilité de la veine porte est recommandée tous les 6 mois par échographie doppler hépatique. L’intérêt d’une anticoagulation en prophylaxie de la TVP est en cours d’évaluation (PHRC APIS). En revanche, le CHC est exceptionnel.
Radiologie interventionnelle
La mise en place d’un TIPS peut être envisagée pour les malades avec hémorragie digestive non contrôlée par les traitements médicaux et endoscopiques. Les résultats sont alors satisfaisants (34). Lorsqu’il existe une ascite réfractaire, la survie après TIPS est plus courte, surtout lorsque la créatininémie est supérieure à 100 µmol/L et/ou lorsqu’il y a des comorbidités importantes (34). Il est important de souligner que compte tenu de l’absence d’insuffisance hépatocellulaire, le score MELD n’est pas adapté pour sélectionner les bons candidats aux TIPS.
Transplantation hépatique
La transplantation hépatique est une option pour les malades avec des complications de l’hypertension portale réfractaires au traitement médical. La survie à long terme rapportée après transplantation hépatique est bonne, avec une survie de 70 % à 5 ans (36). La récidive de la maladie initiale sur le greffon est rare. À l’inverse des lésions de MVPS ont été décrites chez près de 2 % des malades ayant eu une PBH après transplantation hépatique. Cependant, la survenue de manifestations cliniques d’hypertension portale est rare. La pathogénie de ces lésions est mal connue, elles pourraient être liées à un mécanisme de rejet humoral (37, 38).
Autres atteintes des petits vaisseaux du foie
Distension sinusoïdale
La distension sinusoïdale est une lésion histologique définie par une dilatation des sinusoïdes en l’absence d’obstruction des vaisseaux efférents du foie (39). Le diagnostic de distension sinusoïdale primitive ne peut être retenu qu’après avoir écarté un obstacle au retour veineux hépatique (comme une insuffisance cardiaque droite ou péricardite constrictive). Cette lésion est le plus souvent asymptomatique, elle peut être associée à une hépatomégalie. Les douleurs et la fièvre sont exceptionnels. La pathogénie de la distension sinusoïdale est peu claire. Elle a été associée à certains médicaments (en particulier les contraceptifs oraux), des pathologies dysimmunitaires et/ou une inflammation systémique (comme par exemple une infection bactérienne) (40). Même si la biopsie hépatique est l’examen de référence pour le diagnostic, l’aspect en mosaïque du parenchyme hépatique au temps artériel ou portal, qui s’atténue à la phase tardive à la tomodensitométrie (TDM) ou l’imagerie par résonance magnétique (IRM) est fortement évocateur de dilatation sinusoïdale (40). En dehors du traitement étiologique, il n’existe pas de traitement spécifique de la distension sinusoïdale.
Syndrome d’obstruction sinusoïdale (SOS)
Le SOS, anciennement appelé maladie veino-occlusive (MVO), est la perte de l’intégrité de la paroi sinusoïdale, caractérisée par l’obstruction concentrique, non thrombotique, des sinusoïdes et de la lumière des veinules hépatiques, en l’absence de lésion primitive ou de thrombose des veines hépatiques. Le SOS est une complication bien connue des transplantations de cellules souches hématopoïétiques. Dans le contexte du SOS après transplantation de cellules souches hématopoïétiques, les symptômes majeurs du SOS sont la prise de poids (rétention hydro-sodée) avec ou sans ascite, l’hépatomégalie, l’hépatalgie et/ou l’ictère. Il s’agit d’une complication pouvant mettre en jeu le pronostic vital des patients. Le SOS peut également être lié à la toxicité de la chimiothérapie par oxaliplatine, surtout dans le contexte du traitement des métastases hépatiques de cancer colorectal. Il est alors généralement pauci-symptomatique ou entraîne une thrombopénie et une splénomégalie. Cependant, la présence d’un SOS augmente la morbi-mortalité de la chirurgie hépatique (41).
Prise en charge globale des maladies vasculaires du foie
Choix et suivi du traitement anticoagulant
Le traitement par anticoagulant est fréquemment envisagé en cas de maladies vasculaires du foie. Cependant, les recommandations sont de faible niveau de preuve. Compte tenu de la rareté de ces maladies, une décision au cas par cas est indispensable. Elle doit prendre en compte les éléments suivants :
- Le type de maladie ;
- La présence d’un état pro-thrombotique sous-jacent ;
- Les comorbidités ;
- Le risque hémorragique.
L’indication du traitement anticoagulant et le rapport bénéfice/risque doit être réévaluée au moins annuellement.
La présence d’une hypertension portale sévère ne doit pas être considérée comme une contre-indication à un traitement par anticoagulants. En effet, les études suggèrent que lorsqu’une prophylaxie est mise en place, les anticoagulants n’augmentent pas le risque d’hémorragie digestive liée à l’hypertension portale (32, 42, 43). Dans une étude rétrospective portant sur 471 endoscopies chez des malades ayant une TVP chronique non cirrhotique, le maintien des anticoagulants oraux (AVK) n’était pas associé à une augmentation du risque hémorragique (44).
En revanche, le traitement anticoagulant augmente le risque d’hémorragies non liées à l’hypertension portale, notamment avant un geste invasif au cours des maladies vasculaires du foie (42). Ainsi, lorsque cela est possible, un arrêt transitoire du traitement anticoagulant doit systématiquement être discutée avant un geste invasif.
Le traitement anticoagulant de première ligne repose généralement les héparines. Les HBPM doivent être privilégiées, et les héparines non fractionnées doivent être évitées, en raison du risque augmenté de thrombopénie induite à l’héparine, en particulier en cas de syndrome de Budd- Chiari (14). Le traitement est ensuite relayé par un anticoagulant oral. Les antivitamines K ont été largement utilisées dans ce contexte, et restent l’option classique. Compte tenu de leur simplicité, les AOD sont de plus en plus fréquemment prescrits au cours des maladies vasculaires du foie. L’expérience la plus importante avec les AOD concerne le rivaroxaban et l’apixaban. Les données, bien que limitées, suggèrent que les AOD ont une efficacité similaire aux AVK, sans augmentation du risque hémorragique, chez les patients atteints d’hépatopathie chronique (45). Le choix du traitement anticoagulant doit tenir compte de plusieurs facteurs, en particulier de la fonction hépatique, de la fonction rénale, du choix du patient (16). Les AOD ne sont actuellement pas recommandés chez les patients qui ont un SAPL car ils sont associés à un risque accru de thrombose artérielle récurrente (46). Les principales spécificités des anticoagulants oraux sont résumées dans le tableau 3.
Tableau 3 : Principales spécificités des anticoagulants oraux
| Antivitamines K | Rivaroxaban | Apixaban |
| Nécessité d’un monitoring biologique (INR entre 2 et 3) | 1 prise par jour Pas de monitoring | 2 prises par jour Pas de monitoring |
| Difficile à équilibrer en cas d’insuffisance hépatique | Contre-indiqué si insuffisance hépatique modérée (Child Pugh B-C) | Prudence si d’insuffisance hépatique modérée (Child Pugh B) Contre indiqué si insuffisance hépatique sévère (Child Pugh C) |
| Peut être utilisé en cas d’insuffisance rénale | Ne pas utiliser en cas de DFG < 30 ml/min | Ne pas utiliser en cas de DFG < 30 ml/min |
| Données plus limitées, coût plus élevé Pharmacocinétique inconnue après TIPS Non recommandé en cas de syndrome des antiphospholipides | |
TIPS : shunt intrahépatique portosystémique transjugulaire
Mesures associées
Éducation thérapeutique
L’ETP est conçue pour améliorer la qualité de vie et optimiser le soin médical du patient, selon la Haute Autorité de Santé (HAS). L’ETP vise à aider les patients à acquérir ou maintenir les compétences dont ils ont besoin pour gérer au mieux leur vie avec une maladie chronique. Les maladies vasculaires du foie s’intègrent dans cette démarche par le caractère chronique de la pathologie, la prise en charge multidisciplinaire parfois lourde et le recours fréquent à une polymédication notamment les anticoagulants. L’ETP doit être associée à d’autres démarches dans le parcours de soin du patient incluant coordination et prise en compte des traitements concomitants.
Associations de malades
La qualité de vie est fréquemment altérée chez les patients atteints de maladie vasculaire du foie. Il est important d’informer les malades de l’existence des associations de patients dès l’annonce du diagnostic, en l’occurrence l’association des malades des vaisseaux du foie (AMVF, https://www.amvf.asso.fr). La décision d’entrer en relation avec une association reste le choix du patient. Les associations de patients jouent un rôle bénéfique dans le parcours de santé du patient. Elles contribuent à l’accompagnement des malades et de leurs familles en leur apportant des informations sur la pathologie et sa prise en charge, et en luttant contre l’isolement. Elles favorisent les échanges entre les personnes malades et/ou leur famille et apportent un soutien et une aide morale. Leur intervention peut grandement favoriser la compréhension de la pathologie, du traitement et son observance et la communication avec l’équipe médicale.
Conclusion
Les maladies vasculaires du foie représentent un groupe hétérogène de maladies rares. Bien que le diagnostic reste difficile, celui-ci s’est largement amélioré grâce à une meilleure connaissance de ces maladies. Le diagnostic du syndrome de Budd-Chiari repose principalement sur l’imagerie, alors que le diagnostic des maladies des petits vaisseaux du foie nécessite dans la grande majorité des cas une ponction biopsie hépatique. Les maladies vasculaires du foie sont fréquemment associées à des maladies extra-hépatiques, qui doivent être systématiquement recherchées. La prise en charge de ces malades comporte la prise en charge des complications de l’hypertension portale, le traitement des maladies associées. L’organisation en réseau de centres experts (centre de compétence ou de référence maladies rare) a permis de structurer le parcours de soins de ces patients.
Référence
- Jones DEJ, Sturm E, Lohse Access to care in rare liver diseases: New challenges and new opportunities. J Hepatol 2018;68:577–85. https:// doi.org/10.1016/j.jhep.2017.11.004.
- Recommandations AFEF sur les maladies vasculaires du foie – AFEF d. https://afef.asso.fr/recommandation/recommandations-afef-sur-les- maladies-vasculaires-du-foie/ (accessed October 16, 2023).
- Baiges A, Turon F, Simón-Talero M, Tasayco S, Bueno J, Zekrini K, et Congenital Extrahepatic Portosystemic Shunts (Abernethy Malformation): An International Observational Study. Hepatology 2020;71:658–69. https://doi.org/10.1002/hep.30817.
- Garcia-Pagán JC, Valla D-C. Primary Budd-Chiari N Engl J Med 2023;388:1307–16. https://doi.org/10.1056/NEJMra2207738.
- Poisson J, Plessier A, Kiladjian J-J, Turon F, Cassinat B, Andreoli A, et Selective testing for calreticulin gene mutations in patients with splanchnic vein thrombosis: A prospective cohort study. J Hepatol 2017;67:501–7. https://doi.org/10.1016/j.jhep.2017.04.021.
- Magaz M, Alvarez-Larrán A, Colomer D, López-Guerra M, García-Criado MÁ, Mezzano G, et Next-generation sequencing in the diagnosis of non-cirrhotic splanchnic vein thrombosis. J Hepatol 2021;74:89–95. https://doi.org/10.1016/j.jhep.2020.06.045.
- Elkrief L, Payancé A, Plessier A, d’Alteroche L, Ronot M, Paradis V, et Management of splanchnic vein thrombosis. JHEP Rep 2023;5:100667. https://doi.org/10.1016/j.jhepr.2022.100667.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, Fabris F, Eapen CE, Bahr MJ, et al. Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009;151:167–75.
- Ollivier-Hourmand I, Allaire M, Goutte N, Morello R, Chagneau-Derrode C, Goria O, et The epidemiology of Budd-Chiari syndrome in France. Dig Liver Dis 2018;50:931–7. https://doi.org/10.1016/j.dld.2018.04.004.
- Plessier A, Sibert A, Consigny Y, Hakime A, Zappa M, Denninger M-H, et Aiming at minimal invasiveness as a therapeutic strategy for Budd-Chiari syndrome. Hepatology 2006;44:1308–16. https://doi.org/10.1002/hep.21354.
- Seijo S, Plessier A, Hoekstra J, Dell’era A, Mandair D, Rifai K, et Good long-term outcome of Budd-Chiari syndrome with a step-wise management. Hepatology 2013;57:1962–8. https://doi.org/10.1002/hep.26306.
- Plessier A, Esposito-Farèse M, Baiges A, Shukla A, Garcia Pagan JC, De Raucourt E, et Paroxysmal nocturnal hemoglobinuria and vascular liver disease: Eculizumab therapy decreases mortality and thrombotic complications. Am J Hematol 2022;97:431–9. https://doi.org/10.1002/ ajh.26474.
- Desbois AC, Rautou PE, Biard L, Belmatoug N, Wechsler B, Resche-Rigon M, et Behcet’s disease in Budd-Chiari syndrome. Orphanet J Rare Dis 2014;9:104. https://doi.org/10.1186/s13023-014-0153-1.
- Zaman S, Wiebe S, Bernal W, Wendon J, Czuprynska J, Auzinger Increased prevalence of heparin-induced thrombocytopenia in patients with Budd-Chiari syndrome: a retrospective analysis. Eur J Gastroenterol Hepatol 2016;28:967–71. https://doi.org/10.1097/ MEG.0000000000000632.
- Sharma S, Kumar R, Rout G, Gamanagatti SR, Shalimar Dabigatran as an oral anticoagulant in patients with Budd-Chiari syndrome post-percutaneous endovascular intervention. J Gastroenterol Hepatol 2020;35:654–62. https://doi.org/10.1111/jgh.14843.
- Northup PG, Garcia-Pagan JC, Garcia-Tsao G, Intagliata NM, Superina RA, Roberts LN, et al. Vascular Liver Disorders, Portal Vein Thrombosis, and Procedural Bleeding in Patients With Liver Disease: 2020 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2021;73:366–413. https://doi.org/10.1002/hep.31646.
- de Franchis R, Bosch J, Garcia-Tsao G, Reiberger T, Ripoll C, Abraldes JG, et BAVENO VII – RENEWING CONSENSUS IN PORTAL HYPERTENSION. Journal of Hepatology 2021:S0168827821022996. https://doi.org/10.1016/j.jhep.2021.12.022.
- Wang Q, Li K, He C, Yuan X, Luo B, Qi X, et Angioplasty with versus without routine stent placement for Budd-Chiari syndrome: a randomised controlled trial. Lancet Gastroenterol Hepatol 2019;4:686–97. https://doi.org/10.1016/S2468-1253(19)30177-3.
- Garcia-Pagán JC, Heydtmann M, Raffa S, Plessier A, Murad S, Fabris F, et TIPS for Budd-Chiari syndrome: long-term results and prognostics factors in 124 patients. Gastroenterology 2008;135:808–15. https://doi.org/10.1053/j.gastro.2008.05.051.
- Hayek G, Ronot M, Plessier A, Sibert A, Abdel-Rehim M, Zappa M, et Long-term Outcome and Analysis of Dysfunction of Transjugular Intrahepatic Portosystemic Shunt Placement in Chronic Primary Budd-Chiari Syndrome. Radiology 2017;283:280–92. https://doi.org/10.1148/ radiol.2016152641.
- Segev DL, Nguyen GC, Locke JE, Simpkins CE, Montgomery RA, Maley WR, et Twenty years of liver transplantation for Budd-Chiari syndrome: A national registry analysis. Liver Transplantation 2007;13:1285–94. https://doi.org/10.1002/lt.21220.
- Moucari R, Rautou P-E, Cazals-Hatem D, Geara A, Bureau C, Consigny Y, et Hepatocellular carcinoma in Budd-Chiari syndrome: characteristics and risk factors. Gut 2008;57:828–35. https://doi.org/10.1136/gut.2007.139477.
- Van Wettere M, Paulatto L, Raynaud L, Bruno O, Payancé A, Plessier A, et Hepatobiliary MR contrast agents are useful to diagnose hepatocellular carcinoma in patients with Budd-Chiari syndrome. JHEP Rep 2020;2:100097. https://doi.org/10.1016/j.jhepr.2020.100097.
- De Gottardi A, Rautou P-E, Schouten J, Rubbia-Brandt L, Leebeek F, Trebicka J, et Porto-sinusoidal vascular disease: proposal and description of a novel entity. Lancet Gastroenterol Hepatol 2019;4:399–411. https://doi.org/10.1016/S2468-1253(19)30047-0.
- Cazals-Hatem D, Hillaire S, Rudler M, Plessier A, Paradis V, Condat B, et Obliterative portal venopathy: portal hypertension is not always present at diagnosis. J Hepatol 2011;54:455–61. https://doi.org/10.1016/j.jhep.2010.07.038.
- De Gottardi A, Sempoux C, Berzigotti Porto-sinusoidal vascular disorder. J Hepatol 2022;77:1124–35. https://doi.org/10.1016/j. jhep.2022.05.033.
- Barge S, Grando V, Nault J-C, Broudin C, Beaugrand M, Ganne-Carrié N, et al. Prevalence and clinical significance of nodular regenerative hyperplasia in liver biopsies. Liver Int 2016;36:1059–66. https://doi.org/10.1111/liv.12974.
- Guido M, Sarcognato S, Sonzogni A, Lucà MG, Senzolo M, Fagiuoli S, et al. Obliterative portal venopathy without portal hypertension: an underestimate condition. Liver Int 2015.
- Elkrief L, Lazareth M, Chevret S, Paradis V, Magaz M, Blaise L, et Liver Stiffness by Transient Elastography to Detect Porto-Sinusoidal Vascular Liver Disease With Portal Hypertension. Hepatology 2021;74:364–78. https://doi.org/10.1002/hep.31688.
- Valainathan SR, Sartoris R, Elkrief L, Magaz M, Betancourt F, Pellegrino S, et Contrast-enhanced CT and liver surface nodularity for the diagnosis of porto-sinusoidal vascular disorder: A case-control study. Hepatology 2022;76:418–28. https://doi.org/10.1002/hep.32367.
- Wöran K, Semmler G, Jachs M, Simbrunner B, Maria Bauer DJ, Binter T, et al. Clinical Course of Porto-Sinusoidal Vascular Disease Is Distinct From Idiopathic Noncirrhotic Portal Clin Gastroenterol Hepatol 2020:S1542-3565(20)31629-3. https://doi.org/10.1016/j. cgh.2020.11.039.
- Siramolpiwat S, Seijo S, Miquel R, Berzigotti A, Garcia-Criado A, Darnell A, et Idiopathic portal hypertension: natural history and long-term outcome. Hepatology 2014;59:2276–85. https://doi.org/10.1002/hep.26904.
- Schouten JNL, Nevens F, Hansen B, Laleman W, van den Born M, Komuta M, et al. Idiopathic noncirrhotic portal hypertension is associated with poor survival: results of a long-term cohort Aliment Pharmacol Ther 2012;35:1424–33. https://doi.org/10.1111/j.1365-2036.2012.05112.x.
- Bissonnette J, Garcia-Pagán JC, Albillos A, Turon F, Ferreira C, Tellez L, et al. Role of the transjugular intrahepatic portosystemic shunt in the management of severe complications of portal hypertension in idiopathic noncirrhotic portal Hepatology 2016;64:224–31. https://doi. org/10.1002/hep.28547.
- Moga L, Paradis V, Gudavalli K, Silva J, Colecchia A, Ravaioli F, et al. Performance of spleen stiffness measurement by vibration-controlled transient elastography to rule out high-risk varices in patients with porto-sinusoidal vascular Journal of Hepatology 2023;78:S91–2. https:// doi.org/10.1016/S0168-8278(23)00574-3.
- Magaz M, Giudicelli-Lett H, Nicoară-Farcău O, Rajoriya N, Goel A, Raymenants K, et Liver Transplantation for Porto-sinusoidal Vascular Liver Disorder: Long-term Outcome. Transplantation 2023;107:1330–40. https://doi.org/10.1097/TP.0000000000004444.
- Kounis I, Sebagh M, Evain M, Cailliez V, Roche B, De Martin E, et Nodular Regenerative Hyperplasia Is Not a Rare Condition After Liver Transplantation: Incidence, Predictive Factors, and Impact on Survival. Transplantation 2023;107:410–9. https://doi.org/10.1097/ TP.0000000000004303.
- Sebagh M, Yilmaz F, Kounis I, Saliba F, Feray C, Taupin J-L, et Evidence for Alloimmune Sinusoidal Injury in De Novo Nodular Regenerative Hyperplasia After Liver Transplantation. Transpl Int 2023;36:11306. https://doi.org/10.3389/ti.2023.11306.
- Marzano C, Cazals-Hatem D, Rautou P-E, Valla D-C. The significance of nonobstructive sinusoidal dilatation of the liver: Impaired portal perfusion or inflammatory reaction syndrome. Hepatology 2015;62:956–63. https://doi.org/10.1002/hep.27747.
- Ronot M, Kerbaol A, Rautou P-E, Brancatelli G, Bedossa P, Cazals-Hatem D, et Acute extrahepatic infectious or inflammatory diseases are a cause of transient mosaic pattern on CT and MR imaging related to sinusoidal dilatation of the liver. Eur Radiol 2016;26:3094–101. https://doi. org/10.1007/s00330-015-4124-2.
- Aloia T, Sebagh M, Plasse M, Karam V, Lévi F, Giacchetti S, et Liver histology and surgical outcomes after preoperative chemotherapy with fluorouracil plus oxaliplatin in colorectal cancer liver metastases. J Clin Oncol 2006;24:4983–90. https://doi.org/10.1200/JCO.2006.05.8156.
- Rautou P-E, Douarin L, Denninger M-H, Escolano S, Lebrec D, Moreau R, et Bleeding in patients with Budd–Chiari syndrome. Journal of Hepatology 2011;54:56–63. https://doi.org/10.1016/j.jhep.2010.06.019.
- Plessier A, Goria O, Cervoni JP, Ollivier I, Bureau C, Poujol-Robert A, et Rivaroxaban Prophylaxis in Noncirrhotic Portal Vein Thrombosis. NEJM Evidence 2022;1. https://doi.org/10.1056/EVIDoa2200104.
- Guillaume M, Christol C, Plessier A, Corbic M, Péron J-M, Sommet A, et Bleeding risk of variceal band ligation in extrahepatic portal vein obstruction is not increased by oral anticoagulation. Eur J Gastroenterol Hepatol 2018;30:563–8. https://doi.org/10.1097/MEG.0000000000001061.
- Violi F, Vestri A, Menichelli D, Di Rocco A, Pastori D, Pignatelli Direct Oral Anticoagulants in Patients With Atrial Fibrillation and Advanced Liver Disease: An Exploratory Meta-Analysis. Hepatol Commun 2020;4:1034–40. https://doi.org/10.1002/hep4.1513.
- Dufrost V, Wahl D, Zuily Direct oral anticoagulants in antiphospholipid syndrome: Meta-analysis of randomized controlled trials. Autoimmun Rev 2021;20:102711. https://doi.org/10.1016/j.autrev.2020.102711.